
Con frecuencia desentrañamos mecanismos fisiológicos estudiando fenómenos raros, una paradoja que encuentra su ejemplo en las leucodistrofias, enfermedades genéticas de muy baja prevalencia que afectan la mielina, los oligodendrocitos y astrocitos y por las cuales comenzamos a entender su fisiopatogenia relacionada en estos casos al movimiento de agua y solutos.
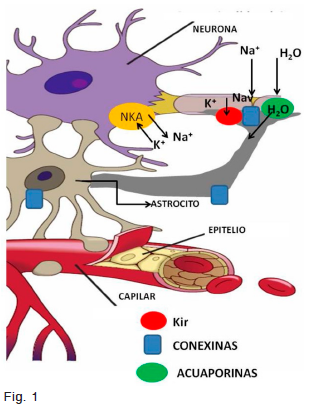
El esquema adjunto simplifica la relación entre un axón mielínico con los astrocitos y por medio de estos con los capilares cerebrales. En la fase de despolarización del potencial de acción, la entrada de Na+ por canales Nav en los nodos de Ranvier es mínima en relación a su concentración intracelular, pero suficiente para provocar un desbalance osmótico que promueve la entrada de agua a la célula. Este exceso de Na+ y agua y la salida periaxonal de K+, en la fase de repolarización del potencial de acción, debe ser balanceada para que la acumulación de K+ en el estrecho espacio extracelular no provoque una despolarización neuronal extendida, como ocurre en las descargas epilépticas1. Para esto se cuenta con dos mecanismos principales, la acción de la NaKATPasa (NKA) y el sincicio de células gliales interconectadas por medio de gap junctions formadas por conexinas (Cx)2, redistribuyendo la concentración local de K+ desde áreas de mayor a otras de menor concentración local, permitiendo que la fuerza impulsora del catión, la diferencia entre el potencial de membrana y el potencial de equilibrio del K+ promueva la entrada del catión hacia el citoplasma por canales Kir. El agua se desplaza por gap junctions también presentes en las láminas de mielina y por las acuaporinas (AQP) (Fig. 1). La estrecha relación entre el
astrocito y el capilar permite que los iones y el agua alcancen el circuito sanguíneo.
En las leucodistrofias y otras raras enfermedades del sistema nervioso existen anormalidades en la expresión de estos mecanismos de transporte. Repasamos algunos ejemplos. Mutaciones recesivas en los genes que codifican las acuaporinas Cx47 subyacen en la enfermedad tipo Pelizaeus-Merzbacher, leucodistrofia con graves anormalidades en el desarrollo motor, funciones cognitivas y espasticidad, en tanto que las que afectan las Cx32 resultan en la enfermedad de Charcot-Marie-Tooth ligada al cromosoma X, neuropatía periférica caracterizada por vacuolización de la mielina y desmielinización. Las mutaciones con pérdida del canal iónico Kir4.1, dan lugar al síndrome SeSAME/EAST, trastorno autosómico recesivo con convulsiones de inicio temprano, sordera neurosensorial, ataxia, retraso mental y desequilibrio de electrolitos por falla renal. El defecto funcional de la NaKATPasa (NKA), por una errata genética en una de sus subunidades, parece constituir la base de la migraña hemipléjica familiar3, 4.
Sí, son enfermedades raras, huérfanas, que pueden ser mortales o debilitantes a largo plazo, de baja prevalencia y alto nivel de complejidad. La mayoría de ellas son enfermedades genéticas; otras son cánceres poco frecuentes, enfermedades autoinmunitarias, malformaciones congénitas, o enfermedades tóxicas e infecciosas. Obligan a los médicos interesados a conocer intricados mecanismos fisiológicos para diagnosticarlas, entenderlas y tratarlas. Conocer su mecanismo es el primer paso para un posible tratamiento. La idea de estudiar estas enfermedades raras fue expresada en el siglo XVII en la carta de William Harvey a un colega, disponible en inglés5 que resumimos así: “En ningún lugar la naturaleza está más acostumbrada a mostrar sus misterios secretos que en los casos en los que muestra rastros de su funcionamiento fuera de los caminos habituales; ni hay mejor manera de promover la práctica de la medicina que una cuidadosa investigación de los casos de las formas más raras de enfermedad”. Chapeaux!
1. Bellot-Saez A, Kékesi O, Morley JW, Buskila Y. Astrocytic mo dulation of neuronal excitability through K(+) spatial buffer – ing. Neurosci Biobehav Rev 2017; 77: 87-97. 2. Chanson M, Kotsias BA, Peracchia C, O’Gra dy SM. Interactions of connexins with other membrane channels and transporters. Prog Biophys Mol Biol 2007; 94: 233-44. 3. van der Knaap MS, Schiffmann R, Mochel F, Wolf NI. Diagnosis, prognosis, and treatment of leukodystrophies. Lancet Neurol 2019; 18: 962-72. 4. Min R, van der Knaap MS. Genetic defects disrupting glial ion and water homeostasis in the brain. Brain Pathol 2018; 28: 372-87. 5. Willis R. The works of William Harvey, MD.1847; 616-7. En: https://www.biodiversitylibrary.org/item/56320#page/7/mode/1up
Comentarios: revmedbuenosaires@gmail.com, kotsias@yahoo.com