
DANIELA FORTUNATI1, JULIO KAPLAN2, JESSICA LÓPEZ MARTÍ3, AGUSTINA PONZONE4, SERGIO INNOCENTI 5, SILVINA FISCINA 4, WALTER CACCIAVILLANO 1, PEDRO ZUBIZARRETA 1, ADRIANA ROSE 1
¹Departamentode Hematología y Oncología, ²Departamento de Diagnóstico por Imágenes, ³Departamentode Patología,
⁴Departamento de Cirugía, 5 Departamento de Ortopedia y Traumatología, Hospital de Pediatría Prof. Dr. Juan P. Garrahan,
Buenos Aires, Argentina
Resumen La fibromatosis tipo desmoide (FD) es un tumor con alta tasa de recurrencia local. Dieciséis pacientes (18 tumores desmoides) fueron evaluados retrospectivamente. La cirugía inicial se realizó en 13/18 tumores, con resección completa en 6 (uno con margen libr e y cinco con margen microscópicamente comprometido); 10/13 tuvieron recaída local. Once pacientes con 13 tumores recibieron tratamiento con metotrexato/vinblastina. La tasa de respuesta a la quimioterapia fue del 54% y de hasta el 81% si se incluyen los casos que lograron enfermedad estable. La mejor respuesta fue remisión parcial. Solo 2 tuvieron toxicidad grado 4. Doce de 15 pacientes tuvieron secuelas. En 8 casos, las secuelas estuvieron directamente relacionadas con la intervención quirúrgica y 3 de ellas fueron graves. La sobrevida libre de progresión a 5 años y la supervivencia global fueron del 30% y del 93.3%, respectivamente. La FD tiene una alta tasa de recaída local, independientemente del margen quirúrgico. Dosis bajas de quimioterapia lograron una enfermedad estable e incluso la remisión de las lesiones, con baja toxicidad. La alta tasa de secuelas probablemente esté relacionada con la cirugía inicial realizada en la mayoría de los pacientes y podría evitarse mediante el uso de quimioterapia neoadyuvante en dosis bajas, como sugieren las estrategias actuales de tratamiento.
Palabras clave: fibromatosis agresiva, fibromatosis tipo desmoide, pacientes pediátricos, cirugía, quimioterapia, secuelas a largo plazo
Abstract Desmoid-type fibromatosis (DF) is a tumor with high local recurrence rate. Sixteen patients (18 desmoid tumors) were retrospectively evaluated. Initial surgery was performed in 13/18 tumors, with complete resection in 6 (one with free margin and five with microscopic residual disease); 10/13 had local relapse. Eleven patients with 13 tumors underwent treatment with methotrexate-vinblastine. The response rate to chemotherapy was 54%, and up to 81% if stable disease cases were included. The best response was partial remission. Only 2 had grade 4 toxicity. Twelve of 15 patients had sequelae. In 8 cases sequelae were directly related to the surgical intervention and 3 of them were severe. The 5-year progression-free survival and overall survival were 30% and 93.3%, respectively. DF has a high local relapse rate, regardless of surgical margin involvement. Low dose chemotherapy achieved stable disease and even remission of the lesions with low toxicity. The high rate of sequelae is probably related to the initial surgery performed in the majority of patients and may be avoided by the use of neoadjuvant low dose chemotherapy.
Key words: aggressive fibromatosis, desmoid-type fibromatosis, surgical treatment, pediatric patients, chemotherapy, long-term sequelae
Postal address: Adriana Rose, Av. Pichincha 1880, 1249 Buenos Aires, Argentina
e-mail: adri.rose27@gmail.com
Desmoid type fibromatosis (DF) is a rare tumor characterized by monoclonal proliferation of fibroblasts in muscles, tendons, and ligaments. DF accounts for 3% of all soft-tissue tumors with an incidence of 0.2-0.4 cases/100 000 inhabitants/year 1. According to the World Health Organization (WHO) 2013 classification of softtissue tumors, DF is considered an intermediate malignant, locally aggressive, and non-metastasizing tumor. Although DF is associated with a high recurrence rate (24-76%) the 10-year overall survival (OS) is higher than 90% 2.
Two forms of presentation have been defined: the sporadic form, more frequently affecting young women, and the form associated with familial adenomatous polyposis (FAP) (7-16%) 3.
Some studies suggest that DF derives from mesenchymal stromal cells 4. A history of trauma is associated with DF 5. Overall, 85% of the sporadic cases have a mutation in the CTNNB1 gene (the β-catenin gene) 6. Some authors have suggested that the mutation spectrum is more complex in children than in adults, associated with AKT (31%), BRAF (19%), and p53 (9%) mutations, in addition to the CTNNB1 gene mutation 7.
DF clinically presents as a firm, often asymptomatic, slow-growing mass, sometimes invading neighboring tissues and compromising vital structures.
Tumor location is divided into abdominal (6-8% of the cases) and extra-abdominal. Children seem to be more affected by head and neck-DF than adults. Head and neck are affected in 26-33% of children with DF, corresponding to the second most prevalent site in this population after primary trunk and limb tumors (versus 7-9% in the adult population)8. A multifocal presentation is less frequently observed 9.
The disease course may be either indolent, in 5-10% of the cases, or aggressive, with a trend towards recurrence despite adequate surgical resection 10. Among favorable prognostic factors, tumor size < 5 cm, head and neck location, and age younger than 10 years have been described 11.
Historically, margin-free surgical resection has been the treatment of choice. However, surgery may be difficult due to the infiltrative growth pattern of the tumor as well as contradictory results regarding microscopic margin status and recurrence-free survival 12. currently it is recommended not to try to achieve margin-negative resection if this results in loss of function or significant morbidity 13-17.
Radiotherapy is limited in children due to the increased risk of second neoplasm and a certain degree of radioresistance (5-year local control of 72% in ≤ 20 years compared to 97% in ≥ 40 years p = 0.009) 18, 19.
Over the last years, low dose chemotherapy (LDC) has been suggested as an alternative to extensive surgical resection 20. Due to the slow-growing nature of the tumor with a slow response to chemotherapy, the treatment should be administered for a prolonged time. LDC consists of a combination of low weekly doses of methotrexate (MTX) together with vinca alkaloids 21. Tamoxifen associated with sulindac or celecoxib is an additional treatment option with a 2-year progression free survival (PFS) rate of 36% 22. A moderate response to hydroxyurea has been reported as well 23.
Despite the heterogeneous behavior of DF, it is well known that the majority of the cases progress within the first two years, with a five-year PFS of 50% without treatment.
Based on these findings, the main international pediatric collaborative groups proposed a conservative (“watch-and-wait”) strategy 21, 24. The aim of this strategy is to evaluate disease behavior, avoiding treatment in patients that will have SD or even disease regression without treatment. If the tumor is located in a site that will compromise organ function, either leading to symptoms or showing rapid progressive growth, LDC should be recommended 25. Primary surgery should only be considered if tumor-free margins can be achieved without sequelae 26.
As previously mentioned, DF is a disease with an excellent survival, being the long-term sequelae, an important issue 27.
The aim of this paper is to describe the clinical characteristics, response to treatment and outcome of a series of pediatric patients with DF treated in a single institution over 14 years, with special emphasis on the analysis of long-term sequelae.
Materials and methods
A retrospective descriptive study was conducted evaluating 18 tumors in 16 patients younger than 16 years with a histological diagnosis of DF treated at the Garrahan Hospital in Buenos Aires, Argentina, between January 2002 and December 2016.
Garrahan Hospital is a third-level pediatric hospital, which treats approximately 40% of pediatric cancer in the country.
Initial tumor size as well as response to treatment was assessed using ultrasonography and/or computer tomography scan, and/or magnetic resonance imaging of the tumor site involved.
From 2005 onwards, patients have been treated according to the guidelines of the EpSSG non RMS 2005 protocol [vinblastine (VBL) 6 mg/m²/week, MTX 30 mg/m²/week], followed by 6 months of spacing the administration to every 2 weeks.
Previously, there were no specific treatment guidelines.
Complete response (CR) was defined as complete disappearance of the tumor, PR as a tumor reduction between 33% and 99%, disease progression as any increase greater than 40%, and SD as cases that did not meet the criteria for either PR or DP. Tumor size was measured in 3 diameters, and tumor volume was determined.
The quality of resection in surgically treated patients was assessed according to the staging system of the Intergroup Rhabdomyosarcoma Study (IRS) 28.
Adverse effects were graded according to the CTC criteria v.4.0 29.
We will refer to the number of tumors, in addition to the number of patients, given that in patients with more than one tumor, these tumors did not have the same evolution.
OS was defined as time from diagnosis to last follow up or death; PFS as the time from diagnosis to progressive disease (PD) or recurrence. Response rate to chemotherapy was defined as the percentage of patients who achieved PR or CR.
OS and PFS curves were calculated with the Kaplan-Meier method using the statistical program IBM SPSS Statistics for Windows, Version 20.0.
Results
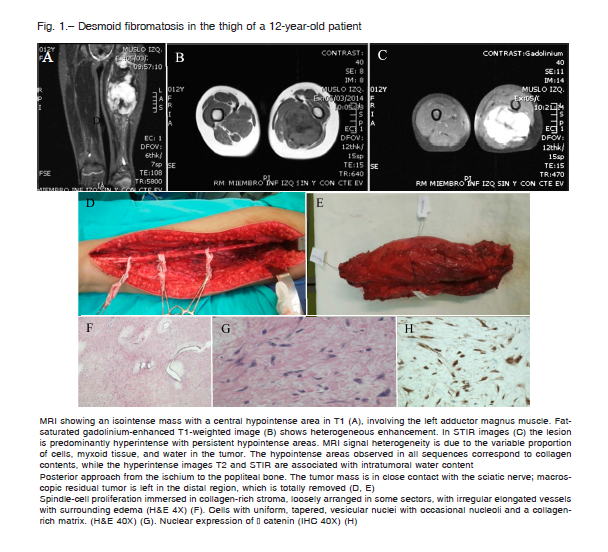
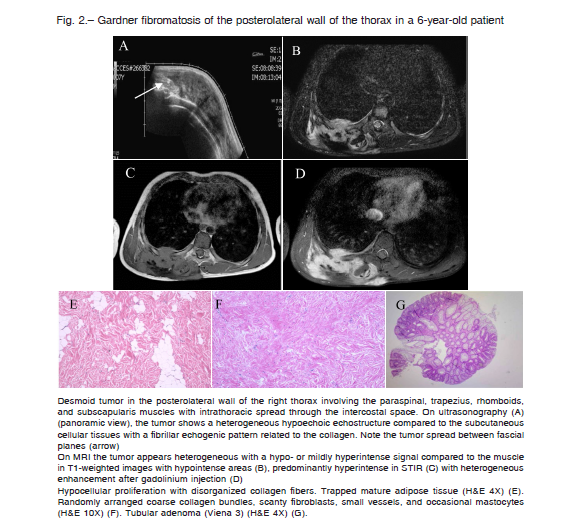
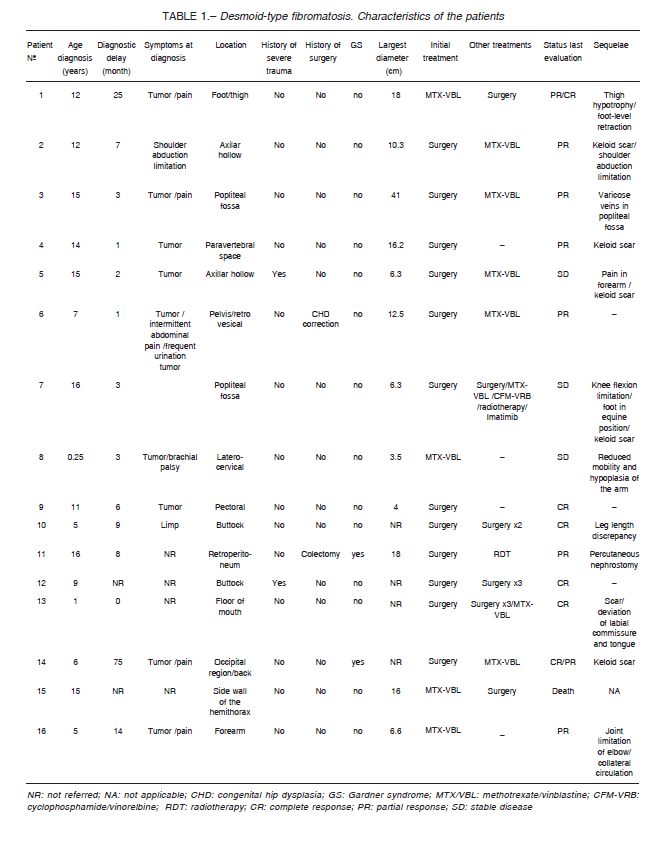
Sixteen patients with 18 desmoid tumors were included in the study. Median age at diagnosis was 11.5 years (range: 0.25-16) and male-to-female ratio was 1:1. The most commonly location was in the limbs (7) and trunk (6), followed by the head and neck (3) and abdomino-pelvic cavity (2) (Fig. 1). Two patients had multifocal tumor, one of whom was associated with FAP. Two patients had a history of surgical trauma, related to correction of congenital dislocation of the hip in one and colectomy in the other, the last related to FAP (Fig. 2). Two patients had a history of trauma, both due to a fall, in one with an associated fracture. Symptoms at diagnosis were tumor (9 patients), pain (5 patients), and decreased range of motion (3 patients). Median largest initial diameter was 9.9 cm (range 3.5-18). In 5 patients nerve involvement and in 3 compressions of adjacent structures was observed.
Median time between symptom onset and biopsy was 358 days (range 28-2282 days). Clinical features of the patients are shown in Table 1.
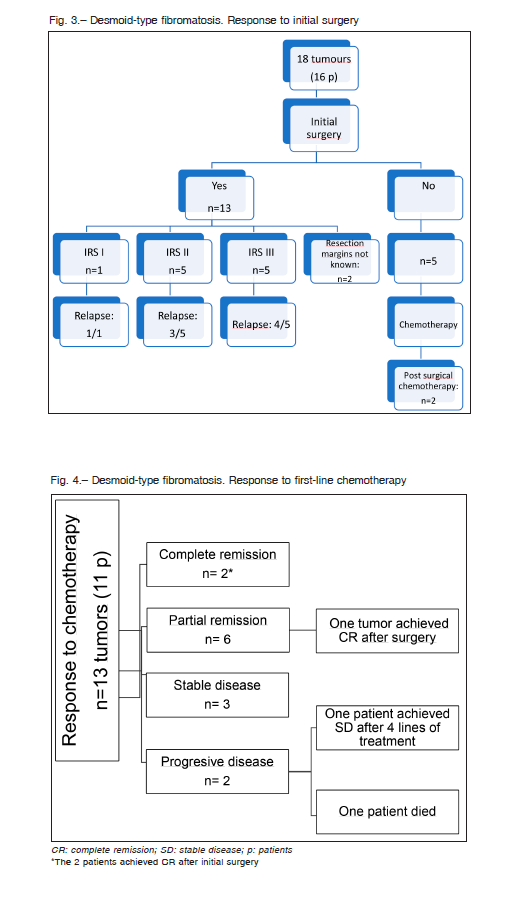
Initial surgery was performed in 13 of 18 tumors (12 patients) with a relapse rate of 76% (10/13), with a median time to relapse of 4.5 months (range 4-9). Four patients achieved CR following surgical treatment alone. Two of the 4 patients required 3 and 4 surgeries, respectively.
Only 2 patients who underwent surgery are alive at the first CR at 12 and 38 months, both were extra-abdominal and with microscopic involvement of surgical resection margin (Fig. 3).
Eleven patients with 13 tumors underwent chemotherapy according to the EpSSG non RMS 2005 protocol. Two patients with 2 tumors each achieved CR in one tumor and PR in the other. In both patients, tumors achieved CR after initial surgery and received chemotherapy as surgical treatment consolidation and to treat the other tumor. The remaining 11 patients (one tumor each) achieved PR in 4 cases, SD in 3 cases and 2 patients presented PD. One of them achieved SD after 4 lines of treatment (MTX/VBL, cyclophosphamide/ vinorelbine, radiotherapy, and imatinib) and the other, who was lost to follow-up 3 months after diagnosis, returned to the hospital with PD and died because of intrathoracic compression of neighboring organs 35 months after the diagnosis. Overall, the response rate to upfront chemotherapy was 54% and up to 81% if SD cases were included (Fig. 4).
Median treatment duration was 11 months (range 1-36). Only 2 patients had significant tumor reduction (at least PR) 3 months after initiation of chemotherapy, while in the remaining patients the reduction occurred after 6 to 15 months. Median time of response to treatment was 8 months (range 1-15).
Two patients were found to have FAP; one of them had congenital multifocal tumor and the other developed a retroperitoneal tumor 12 months post-colectomy. The first patient kept SD for 6 years, until he showed PD, and surgery was performed. After surgery, the tumor showed fast growing and pain, so he underwent metronomic chemotherapy.
The second case underwent surgery followed by radiotherapy and hormonal treatment.
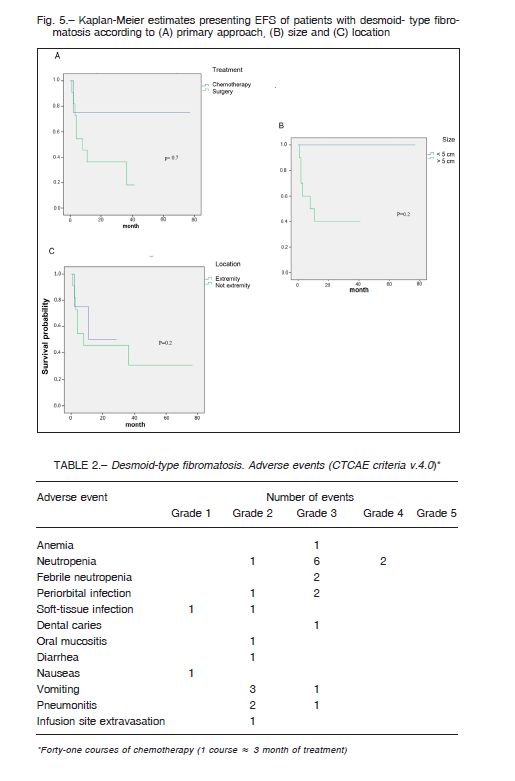
No significant differences in PFS were found when comparing tumor size, location, and initial treatment (Fig. 5).
Only 2 patients had grade 4 toxicity, and neutropenia in both. Out of 41 courses of chemotherapy, febrile neutropenia was observed in two. Seven of 11 patients required a reduction of the VBL dose (of 6 mg/m2 to 4 mg/m2) because of treatment delay due to hematological toxicity (Table 2). After dose reduction there were no new suspensions of the treatment due to toxicity.
Two patients with PD received radiotherapy (dose: 50 -56 Gy) achieving PR in one case (receiving tamoxifen concomitantly with and after radiotherapy) and PD in the other. None of the patients had complications at 12 and 25 months after treatment completion.
Disease status at last evaluation was: one patient died and 15 patients with 17 tumors were alive. Considering tumor status of the last group, 7 tumors were in CR, 7 in PR, and 3 had SD.
Twelve of 15 patients (80%) had sequelae, which were cosmetic in 11, most frequently a keloid scar (5/11), and functional in 9, mainly consisting of decreased range of motion (5/9). In 8 cases sequelae were directly related to the surgical intervention and 3 of them were severe.
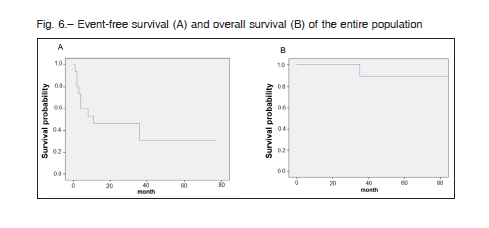
After a median follow-up of 40 months (range 12-149), the 5-year OS and PFS were 93.3% and 30%, respectively (Fig. 6).
Discussion
DF is a rare disease with a variable clinical behavior which has led to a delay in the development of general recommendations for the management of these patients.
Although a number of agents showed activity against desmoid tumors, no accepted standard of care exists for systemic treatment of these tumors 30.
The 5-year PFS and OS in our series were 30% and 93.7%, respectively, similar to the main pediatric series that reported a PFS and OS of 36.1% and 99.4% (Orbach et al.)21, 31% and 88% (Oudot et al) 31, and 44% and 100% (Sparber-Sauer et al)27, respectively, and a PFS of 44% found in the COG study 24.
In our series of 16 patients, the 4 patients treated previously to the year 2005 only underwent surgery and surgery and/or radiotherapy in case of recurrence, while after 2005, treatment with MTX/VBL has been used.
The relapse rate after surgery was 76%, being in the upper limit for the range of recurrence reported in the literature. No correlation was observed between quality of resection and incidence of recurrence which is in line with the results of most recent series 16.
The response rate to chemotherapy was 51%, and up to 81% if SD cases were included. The best response was PR. Although these results are better than those reported by the COG (19%), EpSSG (35%), and Oudot et al (31%), the small sample size of our series makes it difficult to draw definitive conclusions. We should also be careful in attributing a cause-effect relationship to the treatment modalities used, as spontaneous remission has been reported in 20-28% of the cases 32, 33.
Response to treatment was slow, as described in the literature. Nevertheless, some authors reported an early improvement in terms of symptom relief 34. In our series, patients who receive LDC as first-line therapy showed a trend towards a better prognosis than those who underwent initial surgery (this was already described by others); however, this difference was not statistically significant 27. LDC was generally well tolerated, but the first patients in our series required lowering of the VBL dose to 2/3 due to hematological toxicity (in 7 patients).
Neutropenia was the most common adverse effect, as found by other authors 24. No more chemotherapy withdrawal was required after the VBL dose was decreased.
In a recent study conducted in Germany, the initial dose of VBL used was 3 mg/m2, 27.
The only patient who received imatinib achieved disease stabilization after the failure of 3 treatment lines.
Previous studies reported that TKI seem to induce long-term disease stabilization but are associated with low response rates in terms of tumor size reduction.
Imatinib was the first tyrosine kinase inhibitor proposed in DF, showing a response rate of only 6%, however a 1-year PFS of 66% has been reported 35.
Other TKI that have been proposed in DF are pazopaniband and sunitinib 36, 37. Recently, sorafenib has shown promising results with a 2-year event-free survival of 87% and an objective response of 33% versus 36% and 20%, respectively when compared to placebo 38. A recently published review describes common signalling pathways active in DF and provides an up-to-date overview of their therapeutic potential 39.
In our series, the response rate to radiotherapy was 50% which is below the response range reported in the literature; however, due to the small number of patients who underwent the treatment, no conclusions can be drawn. In our patients, the absence of complications may be explained by the short follow-up period, as complications appear after a mean time of 33 months with a rate of 26% after 20 years 40.
The most frequent tumor locations found in our series were in the trunk and limbs, similar to other series; however, primary site in the head and neck was lower than that reported in the literature for DF in children 20.
We did not find statistically significant differences in results regarding to tumor location and size; however, tumors < 5 cm tended to have a better prognosis.
The 2 patients with FAP showed a quite aggressive behavior of the disease and in both cases the surgery acted as a trigger.
Patients with DF and FAP tend to be younger at disease onset and more often have multifocal and intra-abdominal disease 41,42. Surgery is a risk factor for the development of DF in patients with FAP; therefore, in these patients it is recommended to delay prophylactic colectomy 43.
Gardner fibroma has a variable course, with a spectrum ranging from spontaneous regression in approximately 5-10% to a relapsing and remitting course in 30%, SD from onset in 50%, and rapid DP in 10% 44. Optimal management of patients with DF and FAP has not been defined.
Nevertheless, the usefulness of celecoxib, which is also an effective treatment in DF, has been established for advanced adenomas with a decrease in incidence and recurrence 45.
As previously mentioned, DF is a disease with an excellent survival, being the long-term sequelae, an important issue. However, there are only few reports that describe the frequency and nature of them. In the Cooperative Weichteilsarkom Studiengruppe (CWS) publication severe late effects were reported only in 1% of patients who received systemic therapy in contrast to the 20% of patients who were treated by resection(s) 27. A high rate of cosmetic and functional sequelae (80%) was found, and in 20% of patients they were severe. This was probably related to the fact that the majority underwent initial surgical treatment. We have registered a high number of initial surgeries (72% of the tumors), which in the most modern works is discouraged. The reason was that most of the patients were referred from the Orthopedics department after surgery.
In conclusion, DF is a locally aggressive disease with a high local relapse rate, regardless of microscopic involvement of the surgical margins. The outcome of the patients in our series is similar to those reported in the literature.
LDC has allowed to achieved SD and even disease remission with low toxicity with a slow initial but continuous response throughout the treatment. Although the small number of our series and the retrospective nature of the study do not allow us to draw any definitive conclusion, we suggest using lower doses of VBL to avoid the need to delay treatment due to hematologic toxicity.
The high rate of sequelae found in our series is probably related to the initial surgery performed in the majority of patients and may be avoided by the use of neoadjuvant LDC.
Acknowledgement: We are indebted to Dr. Paula Flores, Dr. Lenz Ivonne, Dr. Guido Felizzia and Dr. Marianella Viso for their help in the analysis of clinical and surgical data
Conflicts of interest: None to declare
References
1. Fletcher CDM, Bridge JA, Hogendoorn P, Mertens F (eds). WHO Classification of Tumours of Soft Tissue and Bone, 4th ed. WHO Classification of Tumours, Vol. 5. France: IARC, 2013, p 468.
2. Gi Woon Y, Jae Do K, So Hak C. The analysis of treatment of aggressive fibromatosis using oral methotrexate chemotherapy. Clin Orthop Surg 2014; 6: 439-42.
3. Santos M, Rocha A, Martins V, Santos M. Desmoid tumours in familial adenomatous polyposis: review of 17 patients from a portuguese tertiary center. J Clin Diagn Res 2016; 10: PC01-5.
4. Carothers AM, Rizvi H, Hasson RM, et al. Mesenchymal stromal cell mutations and wound healing contribute to the etiology of desmoid tumors. Cancer Res 2012; 72: 346-55.
5. Cheon SS, Cheah AY, Turley S, et al. Beta-catenin stabilization dysregulates mesenchymal cell proliferation, motility, and invasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proc Natl Acad Sci USA 2002; 99: 6973-8.
6. Kotiligam D, Lazar AJ, Pollock RE, Lev D. Kotiligam D. Desmoid tumor: a disease opportune for molecular insights. Histol Histopathol 2008; 23: 117-26.
7. Meazza C, Belfiore A, Busico A, et al. AKT1 and BRAF mutations in pediatric aggressive fibromatosis. Cancer Med 2016; 5: 1204-13.
8. Paul A, Blouin MJ, Minard-Colin V, et al. Desmoid-type fibromatosis of the head and neck in children: a changing situation. Int J Pediatr Otorhinolaryngol 2019; 123: 33-7.
9. Bekers EM, van Broekhoven DLM, van Dalen T, et al. Multifocal occurrence of extra-abdominal desmoid type fibromatosis – a rare manifestation. a clinicopathological study of 6 sporadic cases and 1 hereditary case. Ann Diagn Pathol 2018; 35: 38-41.
10. Gronchi A, Colombo C, Le Péchoux C, et al. Sporadic desmoid-type fibromatosis: a stepwise approach to a non-metastasising neoplasm–a position paper from the Italian and the French Sarcoma Group. Ann Oncol 2014; 25: 578-83.
11. Risoud M, Mortuaire G, Leroy X, et al. Desmoid tumours of the head and neck in children: review of management. Eur Ann Otorhinolaryngol Head Neck Dis 2017; 134: 155-60.
12. Colombo C, Gronchi A. Desmoid-type fibromatosis: what works best? Eur J Cancer 2009; 45: 466-7.
13. Huang K, Fu H, Shi YQ, Zhou Y, Du CY. Prognostic factors for extra-abdominal and abdominal wall desmoids: a 20-year experience at a single institution. J Surg Oncol 2009; 100: 563-9.
14. Gronchi A, Casali PG, Mariani L, et al. Quality of surgery and outcome in extra-abdominal aggressive fibromatosis: a series of patients surgically treated at a single institution. J Clin Oncol 2003; 21: 1390-7.
15. Bertani E, Testori A, Chiappa A, et al. Recurrence and prognostic factors in patients with aggressive fibromatosis. The role of radical surgery and its limitations. World J Surg Oncol 2012; 10: 184.
16. Woltsche N, Gilg MM, Fraissler L, et al. Is wide resection obsolete for desmoid tumors in children and adolescents? Evaluation of histological margins, immunohistochemical markers, and review of literature. Pediatr Hematol Oncol 2015; 32: 60-9.
17. Grignol VP, Pollock R, Howard JH. Management of desmoids. Surg Clin North Am 2016; 96: 1015-30.
18. Merchant TE, Nguyen D, Walter AW, Pappo AS, Kun LE, Rao BN. Long-term results with radiation therapy for pediatric desmoid tumors. Int J Radiat Oncol Biol Phys 2000; 47: 1267-71.
19. Bates JE, Morris CG, Iovino NM, et al. Radiation therapy for aggressive fibromatosis: the association between local control and age. Int J Radiat Oncol Biol Phys 2018; 100: 997-1003.
20. Meazza C, Bisogno G, Gronchi A, et al. Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer 2010; 116: 233-40.
21. Orbach D, Brennan B, Bisogno G, et al. The EpSSG NRSTS 2005 treatment protocol for desmoid-type fibromatosis in children: an international prospective case series. Lancet Child Adolesc Health 2017; 1: 284-92.
22. Skapek SX, Anderson JR, Hill DA, et al. Safety and efficacy of high-dose tamoxifen and sulindac for desmoid tumor in children: results of a Children’s Oncology Group (COG) Phase II Study. Pediatr Blood Cancer 2013; 60: 1108-12.
23. Ferrari A, Orbach D, Affinita MC, et al. Evidence of hydroxyurea activity in children with pretreated desmoidtype fibromatosis: a new option in the armamentarium of systemic therapies. Pediatr Blood Cancer 2019; 66: e27472.
24. Pounds N, Skapek SX. Desmoid-type fibromatosis in children: a step forward in the cooperative group setting. Am Soc Clin Oncol Educ Book 2012; 593-7.
25. van Broekhoven DL, Grünhagenl DJ, van Dalen T, et al. Tailored beta-catenin mutational approach in extraabdominal sporadic desmoid tumor patients without therapeutic intervention. BMC Cancer 2016; 16: 686.
26. Buitendijk S, van de Ven CP, Dumans TG, et al. Pediatric aggressive fibromatosis: a retrospective analysis of 13 patients and review of the literature. Cancer 2005; 104: 1090-9.
27. Sparber-Sauer M, Seitz G, von Kalle T, et al. Systemic therapy of aggressive fibromatosis in children and adolescents: report of the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer 2018; 65: e26943.
28. Maurer HM, Beltangady M, Gehan EA, et al. The intergroup habdomyosarcoma study-I. A final report. Cancer 1988; 61: 209-20.
29. Common Terminology Criteria for Adverse Events (CTCAE) v4.03. U. S. Department of Health and Human Services. National Institutes of Health. National Cancer Institute. In: https://www.eortc.be/services/doc/ctc/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf; accessed February 2020.
30. Kasper B, Baumgarten C, Garcia J, et al. An update on the management of sporadic desmoid-type fibromatosis: a European Consensus Initiative between Sarcoma Patients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol 2017; 28: 2399-408.
31. Oudot C, Orbach D, Minard-Colin V, et al. Desmoid fibromatosis in pediatric patients: management based on a retrospective analysis of 59 patients and a review of the literature. Sarcoma 2012; 2012: 475202.
32. Fiore M, MacNeill A, Gronchi A, Colombo C. Desmoid-type fibromatosis: evolving treatment standards. Surg Oncol Clin N Am 2016; 25: 803-26.
33. Palassini E, Frezza AM, Mariani L, et al. Long-term efficacy of methotrexate plus Vinblastine/Vinorelbine in a large series of patients affected by desmoid-type fibromatosis. Cancer J 2017; 23: 86-91.
34. Skapek SX, Ferguson WS, Granowetter L, et al. Vinblastine and methotrexate for desmoid fibromatosis in children: results of a Pediatric Oncology Group Phase II Trial. J Clin Oncol 2007; 25: 501-6.
35. Chugh R, Wathen JK, Patel SR, et al. Efficacy of imatinib in aggressive fibromatosis: results of a phase ii multicenter sarcoma alliance for research through collaboration (SARC) trial. Clin Cancer Res 2010; 16: 4884-91.
36. Szucs Z, Messiou C, Wong HH, et al. Pazopanib, a promising option for the treatment of aggressive fibromatosis. Anticancer Drugs 2017; 28: 421-6.
37. Jo JC, Hong YS, Kim KP, et al. A prospective multicenter phase II study of sunitinib in patients with advanced aggressive fibromatosis. Invest New Drugs 2014; 32: 369-76.
38. Gounder MM, Mahoney MR, Van Tine BA, et al. Sorafenib for advanced and refractory desmoid tumors. N Engl J Med 2018; 379: 2417-28.
39. Timbergen MJM, Smits R, Grünhagen DJ, Verhoef C, Sleijfer S, Wiemer EAC. Activated signaling pathways and targeted therapies in desmoid-type fibromatosis: a literature review. Front Oncol 2019; 9: 397.
40. Guadagnolo BA, Zagars GK, Ballo MT. Long-term outcomes for desmoid tumors treated with radiation therapy. Int J Radiat Oncol Biol Phys 2008; 71: 441-7.
41. Koskenvuo L, Peltomäki P, Renkonen-Sinisalo L, et al. Desmoid tumor patients carry an elevated risk of familial adenomatous polyposis: desmoid tumor and the risk of FAP. J Surg Oncol 2016; 113: 209-12.
42. Schäfer M, Kadmon M, Schmidt W, et al. Neonatal gardner fibroma leads to detection of familial adenomatous polyposis: two case reports. European J Pediatr Surg Rep 2016; 4: 17-21.
43. Durno C, Monga N, Bapat B, Berk T, Cohen Z, Gallinger S. Does early colectomy increase desmoid risk in familial adenomatous polyposis? Clin Gastroenterol Hepatol 2007; 5: 1190-4.
44. Cooper K, Squires H, Carroll C, et al. Chemoprevention of colorectal cancer: systematic review and economic evaluation. Health Technol Assess 2010; 14: 1-206.
45. Jung WB, Kim CW, Kim JC. Clinical characteristics and adequate treatment of familial adenomatous polyposis combined with desmoid tumors. Cancer Res Treat 2014; 46: 366-73.