
SILVIA MARINO, NATALIA PEREZ GARRIDO, PABLO RAMÍREZ, MATÍAS PUJANA, GUSTAVO DRATLER, ALICIA BELGOROSKY, ROXANA MARINO
Laboratorio de Pesquisa Neonatal, Laboratorio de Biología Molecular y Diagnóstico, Servicio de Endocrinología, Hospital de Pediatría Prof. Dr. Juan P. Garrahan, Buenos Aires, Argentina
Resumen La hiperplasia suprarrenal congénita (HSC) es un desorden autosómico recesivo producido por la deficiencia de alguna de las enzimas involucradas en la biosíntesis de cortisol. Más del 90% se debe a mutaciones en el gen CYP21A2 que genera deficiencia de 21 hidroxilasa (21OHD). Este gen se encuentra en el brazo corto del cromosoma 6 (6p21·3) y codifica para la enzima citocromo P450C21. Los programas de pesquisa neonatal detectan la forma clásica de la HSC-21OHD cuantificando 17OH-progesterona en gota de sangre en papel de filtro (GSPF). Este test es muy sensible, pero tiene baja especificidad , por lo que se utiliza una segunda muestra para confirmar el resultado. En estos casos, una segunda determinación en la misma muestra podría ser de utilidad. Nuestro objetivo fue evaluar el método de extracción de ADN y posterior análisis molecular del gen CYP21A2 en muestras de GSPF. Analizamos doce individuos presumiblemente afectados por HSC en la pesquisa neonatal usando ADN extraído de sangre fresca recolectada sobre EDTA y de GSPF. Realizamos el análisis del gen CYP21A2 mediante secuenciación automática de todos los exones y regiones intrónicas flanqueantes y MLPA en GSPF, y comparamos los resultados con ambos métodos de extracción. En este estudio demostramos que el ADN extraído de GSPF es una herramienta muy útil para analizar las mutaciones del gen CYP21A2 en la confirmación diagnóstica de 21-OHD para los programas de pesquisa neonatal y que los resultados son comparables con la genotipificación tradicional.
Palabras clave: prueba en gota de sangre seca, hiperplasia suprarrenal congénita, CYP21A2
Abstract Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder due to a deficiency of enzymes involved in cortisol biosynthesis. In more than 90% of cases, CAH is secondary to deleterious mutations in the CYP21A2 gene leading to 21-hydroxilase deficiency (21OHD). The CYP21A2 gene is located on the short arm of chromosome 6 (6p21·3) and encodes the cytochrome P450C21 enzyme. Neonatal screening programs detect the classic forms of CAH-21OHD quantifying 17OH-progesterone in dried blood spots (DBS). This test is very sensitive, but it has a low specificity, requiring a second sample to confirm the result. In these cases, a second-tier test in the same sample may be useful. Our aim was to evaluate a DNA extraction method from DBS and assess the performance of such DNA in the molecular analysis of the CYP21A2 gene mutations. Twelve individuals, who presumably had CAH based on the initial neonatal screening results, were analyzed using DNA extracted from freshly collected blood on EDTA and DBS. The CYP21A2 gene was analyzed by automated sequencing of all exons and intron boundaries and MLPA analysis in DBS. Molecular analysis results from both extraction methods were compared. In this study, we show that DNA extracted from neonatal screening DBS is a useful tool to define CYP21A2 gene mutations in 21-OHD diagnostic confirmation for the newborn screening program and that its results are comparable to traditional genotyping.
Key words: dried blood spot testing, congenital adrenal hyperplasia, CYP21A2
Postal address: Roxana Marino, Servicio de Endocrinología, Hospital de Pediatría Prof. Dr. Juan P. Garrahan, Combate de los Pozos 1881, 1245 Buenos Aires, Argentina
e-mail: marinorox@yahoo.com
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder due to a deficiency of enzymes involved in cortisol biosynthesis. In more than 90% of cases, CAH is secondary to deleterious mutations in the CYP21A2 gene. The CYP21A2 gene is located on the short arm of chromosome 6 (6p21·3) and encodes a cytochrome P450 enzyme (P450C21).
As cortisol synthesis is blocked, ACTH levels increase resulting in overproduction and accumulation of cortisol precursors, particularly 17-OH-progesterone (17OHP).
Dependent on the severity of the disease, 21-OHD is characterized by diminished synthesis of cortisol and
aldosterone and variable degrees of postnatal androgen excess.
Clinically, 21-OHD is classified into classic (including salt-wasting and simple virilizing) and non-classic forms of the disease. In neonates key features of classic 21-OHD are ambiguous genitalia in females, neonatal salt loss, failure to thrive, and potentially fatal hypovolemia and shock. In the following years classic CAH is characterized by rapid postnatal growth, sexual precocity, and different signs of hyperandrogenism. The non-classic form is milder, showing variable degrees of postnatal androgen
excess (hirsutism, menstrual abnormalities) or no clinical symptoms at all (cryptic CAH) 1.
Neonatal screening programs detect the classic forms of CAH by quantifying 17OHP in dried blood spots. This test is very sensitive but it has a low specificity leading to the need for a second sample to confirm the result 2.
Another option may be a second-tier analysis in the same sample consisting of either quantification of 17OHP with organic solvent extraction, a modified liquid chromatography/ tandem mass spectrometry protocol using a ratio of the sum of 17OHP and 21-deoxicortisol levels divided by the cortisol level, or molecular genetics 3.
CYP21A2 mutations can be detected in DNA samples extracted from different sources. Because almost 90% of mutant alleles carry one of 11 mutations, patients carrying none of these mutations are unlikely to be affected. If at least one mutation is detected, the patient is evaluated further. In general, there is a good phenotype-genotype correlation. CYP21A2 genotyping is complicated due to the presence of a nearby-located highly homologous pseudogene CYP21A1P as well as complex duplications, deletions, and rearrangements within chromosome 6p21.3 and thus, requires thorough knowledge on CYP21A2 genetics.
Additionally, CYP21A2 exon and flanking intronic regions sequencing is another option to search for mutations when the 11 most common are not found.
Dried blood spots (DBS) are very convenient since they are minimally invasive. Blood samples are obtained
by pricking the heel or finger, blotted and dried on a filter paper. The advantages include the need for remarkably lower blood volumes and easier shipping and storage at ambient temperatures with good stability of the compounds to be analyzed. This has led to a simplification of the blood collection process and a significant reduction of the costs involved. Additionally, DBS are easy to handle in the laboratory, decrease the risk of transmission of infectious diseases, and represent an inexpensive method for long-term biobanking 4.
DBS samples on filter paper are very useful for genotyping, especially when it is difficult for the patient to travel for a suitable blood extraction, and allows for retrospective study of the disease. Samples can be shipped to centralized laboratories via the standard mail delivery. This type of samples has been used for the detection of mutations in other genes 5, 6 and particularly in the CYP21A2 7-9.
In this study, the DNA extraction method and mutation screening of the CYP21A2 gene from DBS samples were analyzed to be used as another tool in CAH neonatal screening and to compare these results with those obtained from fresh whole blood on EDTA.
Materials and methods
A total of 12 individuals who presumably had CAH based on the initial neonatal screening results were analyzed in this study using DNA extracted from freshly collected blood on EDTA and DNA extracted from DBS.
Additional to these 12 patients assessed to validate the method, 11 other patients were analyzed using DNA extracted from DBS only.
The DELFIA Neonatal 17a-OH-Progesterone kit by Perkin Elmer was used for CAH neonatal screening. It is a competitive solid-phase time-resolved fluoroimmunoassay and values were expressed in nmol/l blood. For the assay, dried blood spot specimens were collected on Whatman 903 specimen collection paper.
Blood samples spotted onto filter paper between 2010 and 2017 were collected from 5 different Newborn Screening laboratories.
All Guthrie Cards were stored at room temperature until molecular analysis.
DNA from DBS on filter paper was extracted from DBS with a commercial extraction kit (QIAamp DNA Mini Kit, Qiagen, Hilden, Germany) according to the manufacturer’s instructions with some modifications.
Briefly, six circles with a diameter of 3 mm each were punched out from DBS. The blood in the six circles was equivalent to 36 μl of whole blood. To increment the amount of DNA extracted, two 50-μl elutions were done. Elution into a fresh tube is recommended to prevent dilution of the first eluate.
DNA was extracted from peripheral blood leucocytes by standard procedures.
Evaluation of DNA quality, detectability and integrity: DNA quality was assessed based on spectrophotometric measurements.
The concentration and absorbance ratio of the DNA were measured at a 260/280 nm using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Massachusetts, United States). DNA detectability was assessed by PCR and DNA integrity by agarose gel 1% electrophoresis.
The 10 most common point mutations in our population were analyzed with allele-specific polymerase chain reaction (PCR), using previously reported primers (p.Ile172Asn, ClEx6, p.Val281Leu, p.Arg356Trp, p.Pro453Ser, p.Arg483ProfsTer58, In2 and Del8bp E3) or RFLP with the PstI restriction enzyme (p.Gln318Ter) and AciI restriction enzyme (p.Pro30Leu) 10, 11. For HGVS recommendations for the nomenclature of the most common mutations at the protein and nucleotide level see footnote Table 2.
When more than one mutation was present in the same patient, molecular studies were performed in both parents, or at least in one if the other was not available, for segregation analysis. To distinguish between homozygosity and hemizygosity, parental samples were analyzed and inferred deletions or large gene conversions were confirmed with MLPA analysis in all cases (MRC Holland P050-CAH version C1, Amsterdam, the Netherlands).
In patients who carried at least one non-detected allele, gene sequence analysis was performed by automated sequencing of all exons and intron boundaries.
Each exon, with flanking intronic regions, was PCR amplified using primers that were previously reported 12. Each purified product (Qia Quick PCR purification kit; Qiagen, Hilden, Germany) was used as a template for direct sequencing using a BigDye Terminator version 3.1 cycle sequencing kit (Applied Biosystems, California, United States) on an ABI PRISM 3130 Genetic Analyzer capillary DNA Sequencer (Applied Biosystems, California, United States).
The nucleotide sequences obtained were compared with the NCBI entry of the CYP21A2 gene: NG_007941.3.
Prior to analysis, written informed consent for the study was obtained from all patients’ parents or tutors. This study was approved by the Ethics Committee of the Garrahan Pediatric Hospital.
In the statistical analysis Fisher’s exact test was used to compare both methodologies for molecular analysis (DNA extracted from DBS vs DNA extracted from fresh blood).
All statistical analyses were performed using EPITABLE Software.
Results
DNA extracted from a total of 12 DBS samples was analyzed.
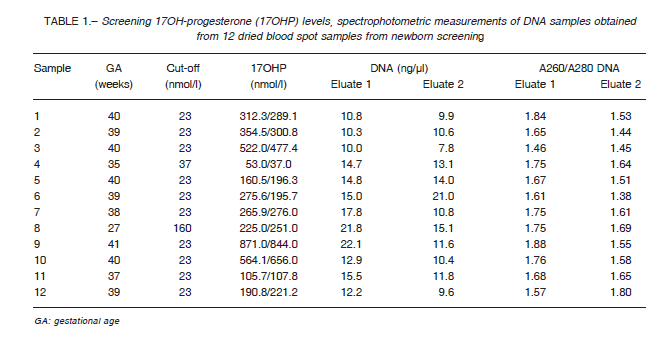
Table 1 shows the evaluation of the quality, detectability, and integrity of the DNA. Mean DNA concentration was 13.10 ± 4.6 ng/ul; 260/280 ratio was found to be 1.72 ± 0.11. DNA integrity as well as detectability by PCR were fairly satisfactory and later sequencing was successful in all the samples.
CYP21A2 mutations can be detected in DNA samples extracted from the same DBS used for hormonal screening.
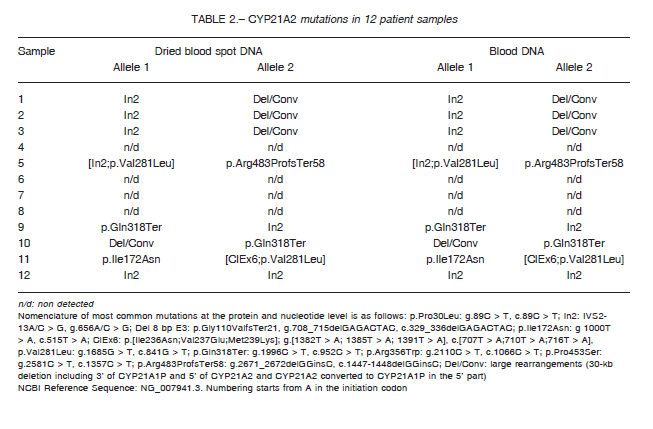
All samples were sequenced and MLPA analysis was performed to detect deletions and gene conversions. In 8 out of 12 patients two affected alleles were detected.
As shown in Table 2, in eight out of 12 samples CAH diagnosis was confirmed, while four patients turned out to be false positive cases, in coincidence with molecular studies performed in DNA extracted from
blood samples.
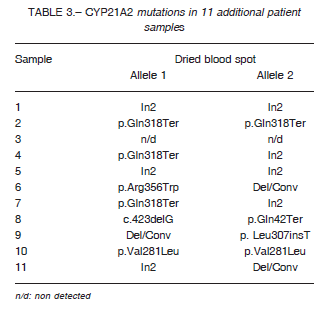
Additionally, we analyzed 11 patients with elevated 17-OHP in neonatal screening using DNA extracted from
DBS only. In these cases, we received DBS from babies who had difficulty coming to the hospital or in whom transportation of the blood sample was complicated enabling molecular study to diagnose CAH.
As shown in Table 3, in 10 out of 11 samples CAH diagnosis was confirmed, while one patient turned out to be a false positive case. The latter was a child born at term with normal weight (gestational age: 39 weeks, weight: 3.980 g) who was admitted to the hospital with severe respiratory distress. He had positive neonatal 17OHP results in two Guthrie cards, so he was referred to a pediatric endocrinologist. The serum sample was lost, but due to the severity of the patient, it was decided to start hydrocortisone and 9-a-fludrocortisone treatment. Once he was compensated and CYP21A2 molecular testing resulted negative, it was decided to reassess and treatment was discontinued. The clinical and endocrinological follow-up confirmed that it was a false positive case.
In the cases in which two mutations were found, analysis of DNA of the parents was necessary to confirm
allele segregation.
To test whether the results of the analysis were reproducible by both methods a Fisher’s exact test was
performed showing that the methods were comparable (one-tailed p = 0.67, two-tailed p = 1.33).
Discussion
In this study, we show that DNA extracted from DBS is a useful tool to define CYP21A2 gene mutations in 21- OHD diagnostic confirmation for the newborn screening program and that its results are comparable to traditional genotyping. The application of this technique allowed to confirm 21-OHD in those patients detected by neonatal screening.
In neonatal screening, careful standardization of the cut-off points is necessary to separate normal babies from those affected with CAH, increasing sensitivity even at the expense of specificity. Determination of 17OHP by fluoroimmunoassay, has a low specificity. In this method, 17OHP is quantitatively displaced from its binding proteins and measured using antibodies supposedly specific to 17OHP. In practice, some cross-reactivity, e.g. with 11-deoxycortisol, could be the cause of false positive values.
Newborns generally have high 17OHP levels in the first two days of life that subsequently decrease, in contrast with those affected with CAH. Females have slightly lower mean 17OHP levels than males. Premature, sick, or stressed infants have higher levels of 17OHP than term infants generating many false positives. Therefore, it is important to use different cut-offs corrected by gestational age or weight at birth.
In addition, if CAH is detected by the screening procedure, differentiation between 21-OHD and 11b-hydroxylase deficiency has to be carried out later on.
The conditions for obtaining and storing the DBS samples are less demanding, improving the accessibility of these tests to centers in less favorable environments.
The diagnostic use of the material obtained from neonatal screening allows shortening of the time needed to make definitive diagnosis.
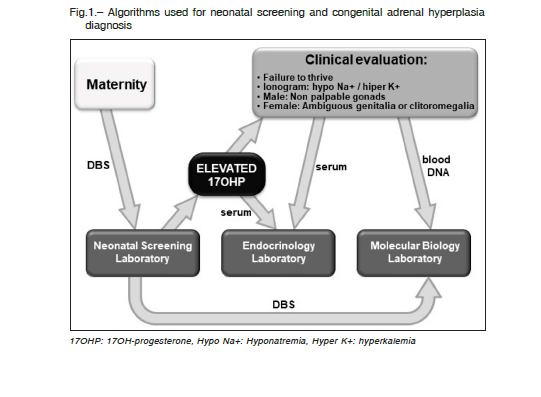
Algorithms currently used for neonatal screening and CAH diagnosis are based on quantification of 17OHP as first tier (Fig. 1). Our results suggest that the same card could be used as a second tier, saving time and money in searching the baby to take a second blood sample.
Early confirmation of the diagnosis by molecular studies without the recitation of a second sample is
important to improve the clinical management of the patient, avoiding salt-wasting crisis and incorrect sex assignment, especially in cases in which it is difficult to contact the patient.
In conclusion, this study confirms the advantage of using DBS as a second tier because it decreases false
positive results that lead to anxiety, saving health-care costs and improving the efficiency of the neonatal screening program.
Conflict of interests: None to declare
References
1. Marino R, Ramirez P, Galeano J, et al. Steroid 21-hydroxylase gene mutational spectrum in 454 Argentinean patients: genotype-phenotype correlation in a large cohort of patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2011; 75: 427-35.
2. Olgemöller B, Roscher AA, Liebl B, Fingerhut R. Screening for congenital adrenal hyperplasia: adjustment of 17-hydroxyprogesterone cutoff values to both age and birth weight markedly improves the predictive value. J Clin Endocrinol Metab 2003; 88: 5790-4.
3. White, P. C. Neonatal screening for congenital adrenal hyperplasia. Nat. Rev. Endocrinol 2009; 5: 490-8.
4. Mark D. Lim. Dried blood Spots for global health diagnostics and surveillance: opportunities and challenges. Am J Trop Med Hyg 2018; 99: 256-65.
5. Sartippour MR, Doroudian R, Frampton G, et al. Identification of galactose-1-phosphate uridyltransferase gene common mutations in dried blood spots. Clin Chim Acta 2014; 436: 298-302.
6. Colon C, Ortolano S, Melcon-Crespo C, et al. Newborn screening for Fabry disease in the north-west of Spain. Eur J Pediatr 2017; 176: 1075-81.
7. Németh S, Riedl S, Kriegshäuser G, et al. Reverse-hybridization assay for rapid detection of common CYP21A2 mutations in dried blood spots from newborns with elevated 17-OH progesterone. Clin Chim Act 2012; 414: 211-4.
8. Malikova J, Votava F, Vrzalova Z, Lebl J, Cinek O. Genetic analysis of the CYP21A2 gene in neonatal dried blood spots from children with transiently elevated 17-hydroxyprogesterone. Clin Endocrinol (Oxf). 2012; 77: 187-94.
9. Kösel S, Burggraf S, Fingerhut R, Dörr HG, Roscher AA, Olgemöller B. Rapid second-tier molecular genetic analysis for congenital adrenal hyperplasia attributable to steroid 21-hydroxylase deficiency. Clin Chem 2005; 51: 298-304.
10. Wedell A., Luthman H. Steroid 21-hydroxylase deficiency: two additional mutations in salt-wasting disease and rapid screening of disease-causing mutations. Human Molecular Genetics 1993; 2: 499-504.
11. Wilson RC, Wei JQ, Cheng KC, Mercado AB, New MI. Rapid deoxyribonucleic acid analysis by allele-specific polymerase chain reaction for detection of mutations in the steroid 21-hydroxylase gene. J Clin Endocrinol Metab 1995; 80: 1635-40.
12. Satomi K, Takio T, Sumitaka S, Kazuhiko S, and Junichi Y. Genetic analysis of Japanese patients with 21-hydroxylase deficiency: identification of a patient with a new mutation of a homozygous deletion of adenine at codon 246 and patients without demonstrable mutations within the structural gene for CYP21. J Clin Endocrinol Metab 2002; 87: 2668-73.
– – – –
In time, a chance will come to reshape our world, prioritise health and wellbeing, work as equal partners with Africa and Asia, and build international solidarity. The agendas of nationalism and isolationism (the ideologies that downgrade public health and primary care), and the selfishness that perpetuates inequalities and a climate disaster, are being destroyed by a pathogen spreading with the ruthless vengeance of a Biblical plague.
Con el tiempo, surgirá la oportunidad de remodelar nuestro mundo, priorizar la salud y el bienestar, trabajar como socios iguales con África y Asia, y construir la solidaridad internacional. Las agendas del nacionalismo y el aislacionismo (las ideologías que degradan la salud pública y la atención primaria), y el egoísmo que perpetúa las desigualdades y un desastre climático, están siendo destruidos por un patógeno que se propaga con la despiadada venganza de una plaga bíblica.
Kamran Abbasi, executive editor
BMJ 2020;369:m1434 doi: 10.1136/bmj.m1434 (Published 9 April 2020)