
JAVIER E. SANCHEZ-SABA 1, MARIANO O. ABREGO 1, JOSÉ I. ALBERGO 1, GERMÁN L. FARFALLI 1, LUIS A. APONTE-TINAO 1, MIGUEL A. AYERZA 1, FEDERICO CAYOL 2, PATRICIA STREITENBERGER 3, MARCELO R. RISK 4, PABLO D. ROITMAN 5
1 Servicio de Ortopedia y Traumatología, 2 Sección Oncología Clínica, Servicio de Clínica Médica, 3 Servicio de Hematooncología Pediátrica, 4 Instituto de Medicina Traslacional e Ingeniería Biomédica, CONICET-Instituto Universitario del Hospital Italiano, 5 Servicio de Anatomía Patológica, Hospital Italiano de Buenos Aires, Argentina
Resumen El sarcoma de Ewing óseo es un tumor poco frecuente, agresivo, que afecta principalmente a niños y adultos jóvenes. Existe ausencia de registros en nuestro país respecto de la prevalencia de esta enfermedad, los esquemas de tratamiento utilizados y sus resultados. El objetivo fue analizar, en un grupo de pacientes con sarcoma de Ewing óseo tratados con quimioterapia y cirugía de conservación de miembro, las tasas de supervivencia global, de recurrencia local y los factores de riesgo oncológicos. Se incluyó a 88 pacientes. La edad media de la serie fue de 14.5 años y el seguimiento promedio de 8.8 años. La tasa de supervivencia global fue de 79.5% a los 2 años, de 69% a 5 años y de 64% a 10 años. Los factores pronósticos negativos asociados a menor supervivencia fueron: mala respuesta a la quimioterapia, edad ≥ de 16 años, localización central, y recurrencia local. En el análisis multivariable únicamente la respuesta a la quimioterapia tuvo significancia estadística. La tasa libre de recurrencia local a 2 y 5 años fue del 87%. La mala respuesta a la quimioterapia fue el único factor significativo para la recurrencia local. Consideramos que la cirugía de conservación de miembro asociada a quimioterapia pre y postoperatoria debe ser el tratamiento para el sarcoma de Ewing óseo, alcanzando de esta manera una supervivencia global a 5 años del 69%. En nuestra serie, la respuesta a la quimioterapia ha sido el factor pronóstico más relevante para supervivencia y recurrencia local.
Palabras clave: sarcoma de Ewing óseo, cirugía de conservación de miembro
Abstract Ewing sarcoma of the bone is a rare, highly aggressive tumor that typically affects children and young adults. In Argentina, the lack of Ewing’s sarcoma registries reflects in the absence of information regarding prevalence, treatment protocols and patient´s outcome. The purpose of this study was to analyze, in a group of patients diagnosed with Ewing sarcoma of the bone, treated with chemotherapy and limb-conserving surgery, their overall survival rate, local recurrence rate, and oncological risk factors. A retrospective research was conducted between 1990 and 2017. Eighty-eight patients with Ewing sarcoma of the bone matched the inclusion criteria. Median age was 14.5 years and median follow-up was 8.8 years. Overall survival rate was 79.5%, 69% and 64% at 2, 5 and 10 years respectively. Negative prognostic factors, associated with less survival rate after univariate analysis, were: bad response to chemotherapy (tumoral necrosis 0-89%), age > 16 years-old, central tumor localization and local recurrence. Gender and tumor size were not significant prognostic factors. After multivariate analysis, response to chemotherapy remained statistical significant. Local recurrence-free survival rate at 2 and 5 years was 87%. Tumor response to chemotherapy (0-89%) was the only significant factor for local recurrence. We consider that limb-salvage surgery, with neoadjuvant and adjuvant chemotherapy, are the mainstays of treatment for Ewing’s sarcoma, with an overall survival rate, at 5 years, of 69%. In this population, response to chemotherapy is the most relevant prognostic factor, being associated with both local recurrence and overall survival.
Key words: Ewing sarcoma of the bond, limb-conserving surgery
Dirección postal: José Ignacio Albergo, Potosí 4032, 1199 Buenos Aires, Argentina
e-mail: nachoalbergo@hotmail.com
El sarcoma de Ewing es una neoplasia poco frecuente, muy agresiva, descrita por primera vez en 1921 por el Dr. James Ewing. Si bien puede localizarse en cualquier parte del cuerpo, se origina con mayor frecuencia en extremidades y pelvis, siendo esta última el sitio más habitual (uno de cada 4 nuevos casos) del sarcoma de Ewing óseo 1-5.
Afecta más frecuentemente a niños y adultos jóvenes, con un pico de incidencia en la segunda década de la vida 3, 5.
El sarcoma de Ewing representa alrededor del 1.5% de todos los cánceres infantiles, y es el segundo tipo más común de sarcoma óseo 6, 7. El diagnóstico de certeza requiere de la evaluación histológica y confirmación con técnicas de citogenética o biología molecular. Al microscopio óptico el sarcoma de Ewing se presenta como un tumor de células pequeñas, redondas y azules, con diferentes patrones arquitecturales y expresión variable de proteínas, detectables mediante técnicas de inmunohistoquímica. La característica que define a estos tumores es la presencia de una serie de translocaciones cromosómicas que culminan en la fusión del gen EWSR1, en el cromosoma 22, con uno de varios miembros de la familia de factores de transcripción ETS. La más común de estas translocaciones, la t (11; 22) (q24; q12), que fusiona el gen EWSR1 con el gen FLI1 en el cromosoma 11, está presente en aproximadamente un 90% de los casos. La detección de estas alteraciones mediante hibridación in situ fluorescente (FISH), RT-PCR o técnicas más modernas como secuenciación de nueva generación (NGS), permiten, junto con la evaluación histológica, un diagnóstico adecuado 8, 9.
La quimioterapia constituye la primera línea de tratamiento. La mayoría de los esquemas utilizan quimioterapia neoadyuvante, seguida de tratamiento local (cirugía) y quimioterapia adyuvante 10. La respuesta histológica a la quimioterapia es posiblemente el indicador pronóstico más importante 11, 12. Sin embargo, a pesar de los cambios en los esquemas de tratamiento en las últimas 3 décadas, la supervivencia global no ha variado significativamente 6, 13-15.
En Argentina son escasos los registros que nos permitan conocer la prevalencia, los esquemas de tratamiento utilizados y valorar los resultados en los pacientes con sarcoma de Ewing. Si nos referimos solo a niños, encontramos datos epidemiológicos concluyentes. Se han registrado 405 casos de sarcoma de Ewing óseo entre los años 2000 y 2016; período en el cual fueron 22 450 los casos de tumores informados en total 16.
El objetivo de este estudio fue analizar un grupo de pacientes con diagnóstico de sarcoma de Ewing óseo tratados con quimioterapia y cirugía de conservación de miembro y determinar tasa de supervivencia global, tasa de recurrencia local y factores de riesgo oncológicos.
Materiales y métodos
Se realizó una búsqueda retrospectiva en nuestra base de datos oncológica entre 1989 y 2017 y se revisaron todas las historias clínicas de los pacientes con diagnóstico de sarcoma de Ewing óseo tratados en nuestra institución. Fueron incluidos aquellos que presentaron un mínimo de un año de seguimiento (pacientes vivos) y que hubieran sido tratados quirúrgicamente. Excluimos aquellos con diagnóstico de sarcoma de Ewing de partes blandas o tratados con radioterapia sin cirugía. Tomamos como punto de inicio de seguimiento el momento del diagnóstico. El punto final se correspondió con la última consulta médica o muerte. El diagnóstico se realizó con biopsias por punción o biopsias quirúrgicas. A partir de 2014, a la histología e inmunohistoquímica, se le sumó la detección de reordenamientos del gen EWSR1 mediante técnica de FISH. Todos los casos fueron examinados por un patólogo especialista en tumores musculoesqueléticos.
El esquema de tratamiento para todos los incluidos en el estudio fue: quimioterapia neoadyuvante + cirugía de resección oncológica + quimioterapia postoperatoria. Dependiendo de los márgenes de resección y respuesta a la quimioterapia se asoció o no radioterapia postoperatoria. Dicha decisión estuvo a cargo de un equipo multidisciplinario y discutido en ateneo. Los esquemas quimioterapéuticos utilizados en niños entre 1990-1999 fueron el esquema VAC (vincristina, actinomicina, ciclofosfamida) y luego se modificó al protocolo VIDE (vincristina, ifosfamida, doxorrubicina, etopósido) desarrollado por la EUROpean Ewing Tumour Working Initiative of National Groups 1999 (Protocolo EURO-E.W.I.N.G. 99) 17.
En adultos los protocolos elegidos fueron: VACA (vincristina, actinomicina, ciclofosfamida doxorrubicina) y VAC/IE (vincristina, ciclofosfamida, doxorrubicina alternando con ifosfamida/etopósido) de acuerdo a estudios fase III 18.
En cuanto a los ciclos de tratamiento quimioterapéutico en pacientes adultos, se realizó un esquema compuesto de 2 ciclos preoperatorios y 5 ciclos postoperatorios. En niños con esquema VAC (1990-1999) se emplearon 4 ciclos preoperatorios y hasta 5 ciclos postoperatorios (un total de 9 ciclos).
En aquellos pacientes tratados bajo el Protocolo EUROE. W.I.N.G. 99 (2000-2017) se realizaron 6 ciclos preoperatorios del esquema VIDE y se completó con 8 ciclos de VAI (vincristina – actinomicina – ifosfamida) de adyuvancia para los localizados. El intervalo entre ciclos es variable dependiendo de la toxicidad y el estado general del paciente.
Todas las piezas de resección fueron procesadas y examinadas según protocolos y recomendaciones del Colegio Americano de Patólogos (CAP) 19. La necrosis se expresó como un porcentaje del volumen total del tumor. Definimos la respuesta al tratamiento de quimioterapia como al porcentaje de necrosis presente luego de la quimioterapia neoadyuvante y resección del tumor primario. La misma fue categorizada como mala (0-89% necrosis) o buena (90-100%).
Se utilizó el método de Kaplan-Meier para analizar la supervivencia global. Se realizó un análisis de regresión logística para las variables dicotómicas: edad (< 16 vs. ≥ 16 años), sexo (masculino vs. femenino), localización primaria del tumor (extremidades vs. central), respuesta a la quimioterapia (mala vs. buena), tamaño tumoral (< 80 mm vs. ≥ 80 mm) y recurrencia local (sí o no). Para el análisis multivariable se utilizaron únicamente las variables con una p < 0.15. Se consideraron valores de p menores a 0.05 como estadísticamente significativos.
Se analizaron las variables para predecir supervivencia de acuerdo con el algoritmo de árboles de decisión20. Todos los análisis fueron desarrollados con el entorno R21.
Resultados
Un total de 88 pacientes fueron incluidos en el estudio. La mediana de edad de la serie al momento del diagnóstico fue de 14.5 años (rango 2-54, percentilo 25%: 9.75 / percentilo 75%: 20 / Diferencia intercuartilos: 10.25) y la mediana de seguimiento fue de 84 meses, (percentilo 25%: 27.75 / percentilo 75%: 145 / Diferencia intercuartilos: 1117.25 de 8.8 años (rango 1-38) (Tabla 1).
La tasa de supervivencia global de la serie analizada fue de 79.5% a los 2 años (IC95%: 71-88), de 69% a 5 años (IC95%:59-79) y de 64% a 10 años (IC95%:57-76) (Fig. 1).
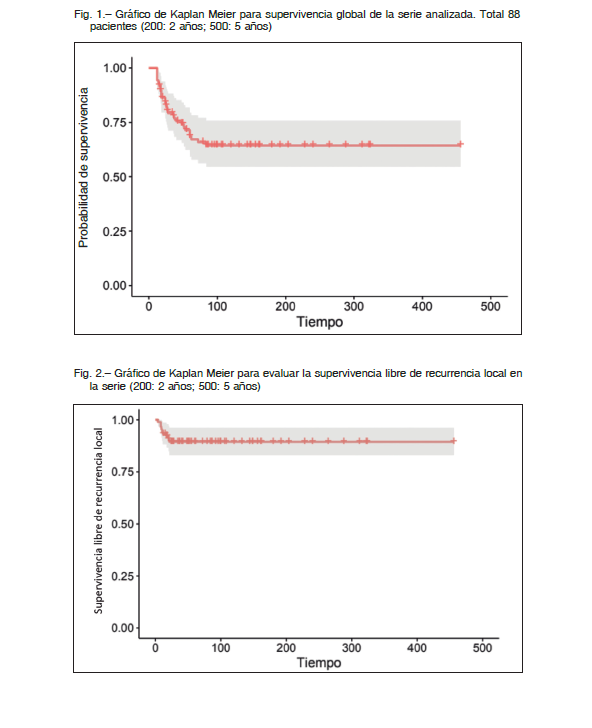
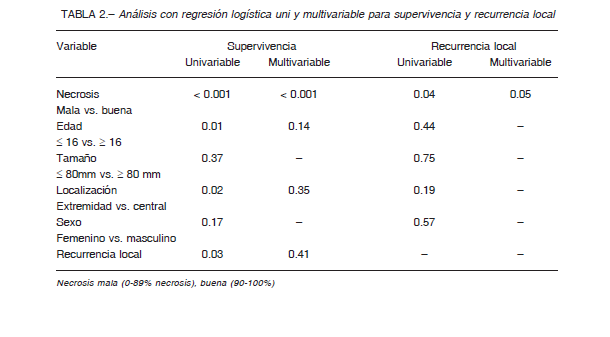
En el análisis de regresión logística univariable, los factores pronósticos negativos asociados en forma estadísticamente significativa a la supervivencia fueron: mala respuesta a la quimioterapia neoadyuvante (0-89%) (p < 0.001/Odds ratio (OR):3.06), edad > a 16 años (p = 0.014/OR: 1.16; localización central (p = 0.02/OR: 1.14) y recurrencia local (p = 0.03/OR: 1.60) (Fig. 2). Ni el sexo ni el tamaño del tumor resultaron ser factores pronósticos significativos (Tabla 2).
En el análisis multivariable, únicamente la respuesta de necrosis tuvo significancia estadística (p < 0.001/OR: 2.99).
Con respecto al control local, se documentaron un total de 7 recurrencias locales en la serie analizada y todas ellas se presentaron antes de los 24 meses postoperatorios.
La tasa libre de recurrencia local a los 2 y 5 años fue del 87% (IC95%:79-97) (Fig. 2). La respuesta a la quimioterapia resultó ser el único factor pronóstico significativo para el desarrollo de una recurrencia local, tanto en el análisis uni como multivariado (p = 0.04 /p = 0.05) (Tabla 2).
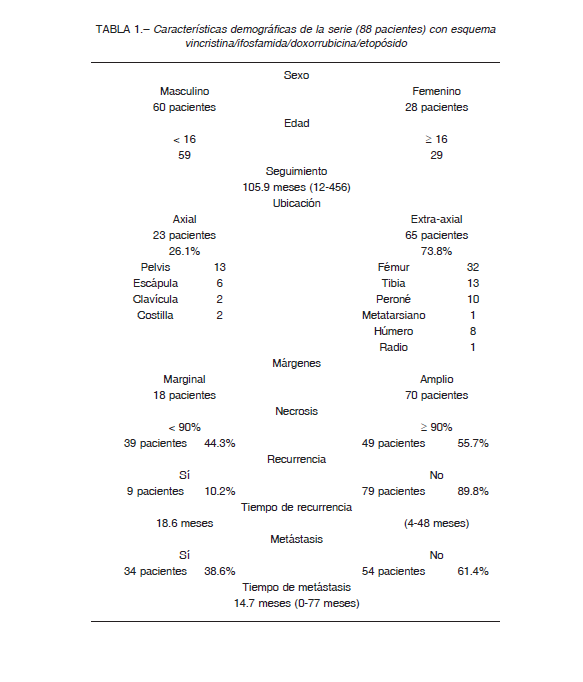
En esta serie de 88 pacientes tratados con cirugía de conservación de miembro no hemos registrado resecciones intralesionales.
Discusión
El sarcoma de Ewing óseo es un tumor poco frecuente. Presenta una incidencia de 1 a 3 nuevos casos por año por cada millón de habitantes en países como EE. UU. y Francia 22. En nuestro país no contamos con datos acerca de su incidencia. El objetivo de nuestro trabajo fue informar los resultados oncológicos de una serie de pacientes con sarcoma de Ewing óseo tratados con quimioterapia y cirugía de conservación de miembro
El tratamiento con quimioterapia es posiblemente el factor más relevante para la supervivencia de los pacientes con sarcoma de Ewing 13, 14, 23. Previo al advenimiento de la quimioterapia, en el sarcoma de Ewing se informaban tasas de supervivencia muy bajas (5-10% a dos años) a pesar de los tratamientos quirúrgicos agresivos 24.
Los esquemas terapéuticos multimodales (quimioterapia, cirugía y/o radioterapia) incrementaron la supervivencia a 5 años en enfermedad localizada a más del 65% 6, 25-27. En nuestra serie, la supervivencia global a 5 años fue de 69% (IC95%:59-79).
A pesar de los avances terapéuticos, los pacientes con sarcoma de Ewing metastásico presentan una supervivencia a 5 años que no supera el 35%. Se ha informado que hasta 40% de los pacientes con sarcoma de Ewing óseo pueden desarrollar una recurrencia local. Las tasas de supervivencia posteriores a la recurrencia de la enfermedad son bajas, con valores que oscilan entre el 15% y 25% a 5 años post recaída 28, 29. En nuestra serie, hemos registrado una tasa de recurrencia local de 7.8% a 2 años y asociación entre recurrencia local y pobre pronóstico oncológico (p = 0.03). El control local parece ser determinante para la supervivencia.
En la década del 70, la introducción de la doxorrubicina al ya conocido esquema tradicional compuesto por vincristina, actinomicina y ciclofosfamida (VAC); así como el agregado de ifosfamida/etopósido (IE) al también tradicional esquema compuesto por vincristina, doxorubicina y ciclofosfamida (VDC) demostraron mejores tasas de supervivencia libre de eventos en pacientes con sarcoma de Ewing óseo no metastásico 14, 17, 30. En nuestra serie, a partir del año 2000 todos los niños fueron tratados con el esquema VIDE, surgido luego del protocolo EURO-E.W.I.N.G. 99. Sin embargo, en los adultos, no hemos podido homogeneizar el tratamiento quimioterapéutico, lo que dificulta el análisis en detalle de este grupo de pacientes en particular, resultando en una limitación del trabajo.
La respuesta histológica a la neoadyuvancia fue descrita como uno de los factores pronósticos más relevantes 31-34. Un 90% de necrosis es el punto de corte habitual para establecer si el paciente es buen o mal respondedor a la quimioterapia neoadyuvante 11. Recientemente se estableció que aquellos con un 100% de necrosis en la pieza quirúrgica presentan mejor supervivencia global en comparación incluso con aquellos con 99% de necrosis.
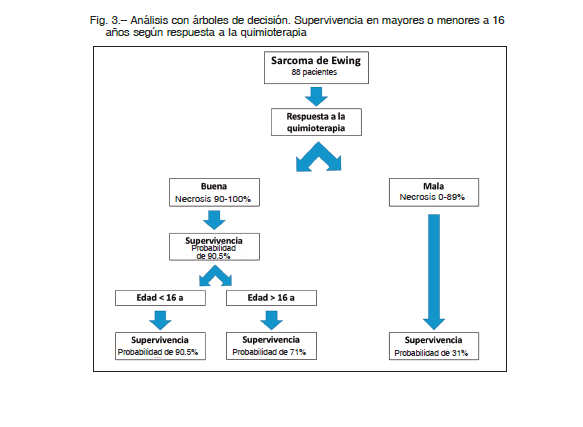
Este cambio de paradigma implica nuevos valores de corte en donde únicamente los que tienen 100% de necrosis deberían ser considerados como excelentes respondedores 12. Debido al número de pacientes de nuestra serie, el análisis de respuesta a la quimioterapia debió hacerse de manera dicotómica (mala vs. buena), poniendo el valor de corte en 90%. Sin lugar a dudas, se trata de una limitación de nuestro estudio, que esperamos poder solucionar con trabajos nacionales multicéntricos que permitan aumentar el número de casos. En nuestra serie, aquellos con porcentajes de necrosis menores a 90% presentaron un pronóstico significativamente peor (p < 0.001) (Fig. 3). Si bien reiteramos que hoy en día solo aquellos con 100% de necrosis deberían ser considerados como excelentes respondedores, en nuestro estudio una necrosis tumoral por encima del 90% se asoció a una supervivencia mucho más alta, del 90.5%
Recientemente, Bosma y col. 35 llevaron a cabo una revisión sistemática con el fin de evaluar factores pronósticos para la supervivencia en sarcoma de Ewing. Dentro de las variables analizadas se incluyeron el tamaño y el volumen tumoral. Si bien el poder estadístico no alcanza para afirmarlo, un diámetro tumoral de más de 80 mm se relaciona con un descenso en la supervivencia global.
Contrariamente, en nuestra serie el tamaño tumoral no se asoció a una peor supervivencia. Independientemente de las dimensiones del tumor, hoy sabemos que el control local de la enfermedad mediante resección quirúrgica tumoral con márgenes libres forma parte del estándar de oro para el tratamiento del sarcoma de Ewing. Los márgenes quirúrgicos demostraron ser un factor pronóstico de relevancia tanto para la supervivencia global como para el control local. Existe asociación entre la presencia de márgenes quirúrgicos amplios y supervivencia global. Es más, márgenes histológicos mayores a 1 cm impresionan ser mejores en términos oncológicos que las resecciones marginales (0.1-1cm) 34-36.
La asociación del tratamiento quirúrgico con radioterapia local parece ser hoy la mejor indicación en sarcoma de Ewing, especialmente en los de localización central, con márgenes contaminados o en aquellos con mala respuesta a la quimioterapia. 37, 39-42. Ya en 2003, Bacci y col.34 establecieron que en aquellos casos con márgenes quirúrgicos inadecuados, es necesaria la radioterapia postoperatoria. Así como la quimioterapia tiene como objetivo principal controlar la diseminación de la enfermedad, la radioterapia juega un rol importante en el control de la recurrencia local 43. Una de las indicaciones de radioterapia postoperatoria es la presencia de márgenes quirúrgicos positivos. A su vez, se recomienda utilizar radioterapia en casos en los cuales la resección quirúrgica sea menor en relación al área afectada por el tamaño tumoral prequimioterapia 44.
Los protocolos actuales sobre sarcoma de Ewing incluyen a la mala respuesta a la quimioterapia, como un factor para indicar radioterapia postoperatoria 45.
En tumores de localización axial (pelvis, columna vertebral, parrilla costal) se sugiere el empleo de radioterapia postoperatoria debido a la dificultad que conlleva realizar resecciones quirúrgicas amplias resguardando estructuras nobles 45-47.
En 2017, Verma y col. 42 realizaron un estudio retrospectivo comparativo incluyendo 1870 pacientes (976 niños y 894 adultos) con sarcoma de Ewing; concluyendo que el grupo de adultos presentaba una menor supervivencia global que el grupo de niños. En concordancia con lo mencionado; en nuestra serie los menores de 16 años (pediátricos) presentaron mejores tasas de supervivencia a 5 años (78% vs. 54%/p = 0.014), que los adultos.
El artículo nacional más relevante, el cual puede encontrarse en la base de datos más importante de la literatura anglosajona, es aquel publicado por Schwartz y col. 48 en 1990. Ya en aquel entonces los pacientes con sarcoma de Ewing eran tratados mediante esquemas que combinaban cirugía, radioterapia y quimioterapia.
Nuestro estudio presenta ciertas limitaciones: el diseño metodológico retrospectivo; el amplio período de análisis (20 años), con las diferencias en técnicas quirúrgicas utilizadas, material de osteosíntesis o quirófano (con o sin flujo laminar); la carencia de un análisis de los resultados en términos de los diferentes esquemas de quimioterapia y la falta de datos acerca del tratamiento asociado con radioterapia, lo que no nos permitió un análisis detallado de esta variable. Además, cabe mencionar que el presente no es un estudio multicéntrico, lo cual implica una limitante a la hora de extrapolarlo a nivel nacional. Sin embargo, nuestra institución es un centro de derivación de la enfermedad y el número de pacientes incluidos en este estudio es el más amplio a nivel nacional.
Consideramos que la cirugía de conservación de miembro asociada a quimioterapia pre y postoperatoria constituye el tratamiento para el sarcoma de Ewing óseo, alcanzando una supervivencia global a 5 años del 69%. La respuesta a la quimioterapia es el factor pronóstico más relevante, asociado tanto a recurrencia local como a la supervivencia.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Ewing J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York Pathological Society, 1921. CA Cancer J Clin 1972; 22: 95-8.
2. Glass AG, Fraumeni JF. Epidemiology of bone cancer in children. J Natl Cancer Inst 1970; 44: 187-99.
3. Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol 2000; 18: 3108-14.
4. Widhe B, Widhe T. Initial symptoms and clinical features in osteosarcoma and Ewing sarcoma. J Bone Joint Surg Am 2000; 82: 667-74.
5. Whelan J, McTiernan A, Cooper N, et al. Incidence and survival of malignant bone sarcomas in England 1979-2007. Int J Cancer 2012; 131: E508-17.
6. Esiashvili N, Goodman M, Marcus RB. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 2008; 30: 425-30.
7. Price CH, Jeffree GM. Incidence of bone sarcoma in SW England, 1946-74, in relation to age, sex, tumour site and histology. Br J Cancer 1977; 36: 511-22.
8. Zucman J, Melot T, Desmaze C, et al. Combinatorial generation of variable fusion proteins in the Ewing family of tumours. EMBO J 1993; 12: 4481-7.
9. Peter M, Couturier J, Pacquement H, et al. A new member of the ETS family fused to EWS in Ewing tumors. Oncogene 1997; 14: 1159-64.
10. Rodriguez-Galindo C. Pharmacological management of Ewing sarcoma family of tumours. Expert Opin Pharmacother 2004; 5: 1257-70.
11. Paulussen M, Craft AW, Lewis I, et al. Results of the EICESS-92 Study: two randomized trials of Ewing’s sarcoma treatment–cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in highrisk patients. J Clin Oncol 2008; 26: 4385-93.
12. Albergo JI, Gaston CL, Laitinen M, et al. Ewing’s sarcoma: only patients with 100% of necrosis after chemotherapy should be classified as having a good response. Bone Joint J 2016; 98-B: 1138-44.
13. Kushner BH, Meyers PA, Gerald WL, et al. Very-high-dose short-term chemotherapy for poor-risk peripheral primitive neuroectodermal tumors, including Ewing’s sarcoma, in children and young adults. J Clin Oncol 1995; 13: 2796-804.
14. Nesbit ME, Gehan EA, Burgert EO, et al. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the First Intergroup study. J Clin Oncol 1990; 8: 1664-74.
15. Sauer R, Jürgens H, Burgers JM, Dunst J, Hawlicek R, Michaelis J. Prognostic factors in the treatment of Ewing’s sarcoma. The Ewing’s Sarcoma Study Group of the German Society of Paediatric Oncology CESS 81. Radiother Oncol 1987; 10:101-10.
16. Registro Oncopediátrico Hospitalario Argentino. En: http://www.msal.gov.ar/inc/epidemiologia-y-registros/roha/; consultado septiembre de 2018.
17. Juergens C, Weston C, Lewis I, et al. Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer 2006; 47: 22-9.
18. Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003; 348: 694-701.
19. Rubin BP, Antonescu CR, Gannon FH, et al. Protocol for the examination of specimens from patients with tumors of bone. Arch Pathol Lab Med 2010; 134: e1-7.
20. Podgorelec V, Kokol P, Stiglic B, Rozman I. Decision trees: an overview and their use in medicine. J Med Syst 2002; 26: 445-63.
21. R Core Team. 2018. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. R Foundation for Statistical Computing, Vienna. En: https://www.r-project.org; consultado septiembre 2018.
22. Tirode F, Surdez D, Ma X, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 2014; 4: 1342-53.
23. Craft AW, Cotterill SJ, Bullimore JA, Pearson D. Long-term results from the first UKCCSG Ewing’s Tumour Study (ET-1). United Kingdom Children’s Cancer Study Group (UKCCSG) and the Medical Research Council Bone Sarcoma Working Party. Eur J Cancer 1997; 33: 1061-9.
24. Dahlin DC, Coventry MB, Scanlon PW. Ewing’s sarcoma. A critical analysis of 165 cases. J Bone Joint Surg Am 1961; 43-A: 185-92.
25. Damron TA, Ward WG, Stewart A. Osteosarcoma, chondrosarcoma, and Ewing’s sarcoma: National Cancer Data Base Report. Clin Orthop Relat Res 2007; 459: 40-7.
26. Gaspar N, Hawkins DS, Dirksen U, et al. Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol 2015; 33:3036-46.
27. Werier J, Yao X, Caudrelier J-M, et al. A systematic review of optimal treatment strategies for localized Ewing’s sarcoma of bone after neo-adjuvant chemotherapy. Surg Oncol 2016; 25: 16-23.
28. Bacci G, Ferrari S, Longhi A, et al. Therapy and survival after recurrence of Ewing’s tumors: the Rizzoli experience in 195 patients treated with adjuvant and neoadjuvant chemotherapy from 1979 to 1997. Ann Oncol 2003; 14: 1654-9.
29. Stahl M, Ranft A, Paulussen M, et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 2011; 57: 549-53.
30. Yock TI, Krailo M, Fryer CJ, et al. Local control in pelvic Ewing sarcoma: analysis from INT-0091–a report from the Children’s Oncology Group. J Clin Oncol 2006; 24: 3838-43.
31. Le Deley M-C, Paulussen M, Lewis I, et al. Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J Clin Oncol 2014; 32: 2440-8.
32. Picci P, Rougraff BT, Bacci G, et al. Prognostic significance of histopathologic response to chemotherapy in nonmetastatic Ewing’s sarcoma of the extremities. J Clin Oncol 1993; 11: 1763-9.
33. Picci P, Böhling T, Bacci G, et al. Chemotherapy-induced tumor necrosis as a prognostic factor in localized Ewing’s sarcoma of the extremities. J Clin Oncol 1997; 15:1553-9.
34. Bacci G, Ferrari S, Bertoni F, et al. Prognostic factors in nonmetastatic Ewing’s sarcoma of bone treated with adjuvant chemotherapy: analysis of 359 patients at the Istituto Ortopedico Rizzoli. J Clin Oncol 2000; 18: 4-11.
35. Bosma SE, Ayu O, Fiocco M, Gelderblom H, Dijkstra PDS. Prognostic factors for survival in Ewing sarcoma: A systematic review. Surg Oncol 2018; 27: 603-10.
36. Bacci G, Ferrari S, Longhi A, et al. Role of surgery in local treatment of Ewing’s sarcoma of the extremities in patients undergoing adjuvant and neoadjuvant chemotherapy. Oncol Rep 2004; 11: 111-20.
37. Arpaci E, Yetisyigit T, Seker M, et al. Prognostic factors and clinical outcome of patients with Ewing’s sarcoma family of tumors in adults: multicentric study of the Anatolian Society of Medical Oncology. Med Oncol 2013; 30: 469.
38. Miller BJ, Gao Y, Duchman KR. Does surgery or radiation provide the best overall survival in Ewing’s sarcoma? A review of the National Cancer Data Base. J Surg Oncol 2017; 116: 384-90.
39. Rodríguez-Galindo C, Liu T, Krasin MJ, et al. Analysis of prognostic factors in ewing sarcoma family of tumors: review of St. Jude Children’s Research Hospital studies. Cancer 2007; 110: 375-84.
40. Lee J, Hoang BH, Ziogas A, Zell JA. Analysis of prognostic factors in Ewing sarcoma using a population-based cancer registry. Cancer 2010; 116: 1964-73.
41. Biswas B, Rastogi S, Khan SA, et al. Developing a prognostic model for localized Ewing sarcoma family of tumors: A single institutional experience of 224 cases treated with uniform chemotherapy protocol. J Surg Oncol 2015; 111: 683-9.
42. Verma V, Denniston KA, Lin CJ, Lin C. A Comparison of Pediatric vs. Adult Patients with the Ewing Sarcoma Family of Tumors. Front Oncol 2017; 7: 82.
43. Sluga M, Windhager R, Lang S, et al. The role of surgery and resection margins in the treatment of Ewing’s sarcoma. Clin Orthop Relat Res 2001; 392: 394-9.
44. Foulon S, Brennan B, Gaspar N, et al. Can postoperative radiotherapy be omitted in localised standard-risk Ewing sarcoma? An observational study of the Euro-E.W.I.N.G group. Eur J Cancer 2016; 61: 128-36.
45. Albergo JI, Gaston CLL, Parry MC, et al. Risk analysis factors for local recurrence in Ewing’s sarcoma: when should adjuvant radiotherapy be administered? Bone Joint J 2018; 100-B: 247-55.
46. Vogin G, Helfre S, Glorion C, et al. Local control and sequelae in localised Ewing tumours of the spine: a French retrospective study. Eur J Cancer 2013; 49: 1314-23.
47. Werier J, Yao X, Caudrelier J-M, et al. Evidence-based guideline recommendations on treatment strategies for localized Ewing’s sarcoma of bone following neo-adjuvant chemotherapy. Surg Oncol 2016; 25: 92-7.
48. Schwartz L, Kupferman J, Picco P. Segundo tumor maligno en pediatría. Comunicación de 22 casos. Medicina (B Aires) 1990; 50: 97-101.