
FLORENCIA AGUIRRE, ANDRÉS M. VILLA
División Neurología, Hospital General de Agudos Dr. José María Ramos Mejía, Centro Argentino de Neuroinmunología, CADENI, Facultad de Medicina, Universidad de Buenos Aires, Argentina
Resumen La miastenia gravis (MG) es una enfermedad autoinmune mediada por anticuerpos dirigidos contra proteínas post sinápticas de la unión neuromuscular. El objetivo de este estudio fue describir los aspectos clínicos, epidemiológicos y serológicos de pacientes con MG en un Hospital Público de la Ciudad de Buenos Aires. Se realizó un análisis retrospectivo sobre 190 enfermos con diagnóstico de MG. La edad media de inicio de la enfermedad fue de 38 años; 57 (30%) fueron MG de inicio tardío (inicio de síntomas > 50 años). La relación mujer/hombre fue 1.7/1. La enfermedad se inició más tempranamente en las mujeres que en los hombres, media 32 vs. 48 años (p < 0.0001). La MG familiar autoinmune representó el 3.2 % (6 casos). La forma más común de presentación fue con manifestaciones oculares puras (52%). El 12.1% (23/190) fue considerada MG ocular en el seguimiento. La MG asociada a timoma se presentó en 22 casos (11.6%). El 27.1% presentó otra enfermedad autoinmune asociada, siendo las tiroideas las más frecuentes. El 81.4% tuvo anticuerpos antireceptores de acetilcolina (ACRA) positivos y 22.7% de los ACRA negativos fueron positivos para anticuerpos anti-tirosina quinasa musculo especifica (anti-MusK). La evolución clínica fue favorable, hallándose más de la mitad de los casos en remisión o manifestaciones mínimas en la última visita. La mayoría requirió inmunosupresión para control de la sintomatología, el 78% recibió corticoides y el 48% un inmunosupresor no esteroideo.
Palabras clave: miastenia gravis, anticuerpos, aspectos clínicos, evolución, epidemiología
Abstract Myasthenia gravis (MG) is an antibody-mediated autoimmune disease of the neuromuscular junction. The aim of this study was to evaluate clinical, epidemiological and serological features of patients with MG in a Public Hospital of Buenos Aires City. A retrospective analysis of 190 patients diagnosed with MG was performed. The mean age of MG onset was 38 years, 30% had late-onset MG (onset age > 50 years). The female/male ratio was 1.7 / 1. Disease started earlier in women than in men, mean 32 vs. 48 years (p < 0.0001). Familial autoimmune MG represented 3.2% of the cases. Most of the patients initiated their disease with a pure ocular form (52%). 12.1% (23/190) were considered ocular MG at follow-up. Thymoma-associated MG represented 11.6% of cases. 27.1% had other associated autoimmune disease, thyroid disorders were the most frequent. 81.4% were anti-acetylcholine receptor antibody (AChR-ab) positive MG; 22.7% of AChR-ab negatives were positive for anti-muscle specific kinase (MusK) antibodies. Clinical outcome was relatively good; more than half of cases were in remission or minimal manifestations at the last visit. The majority of patients required immunosuppression to control the symptoms, 78% received corticosteroids and 48%, a non-steroidal immunosuppressant.
Key words: myasthenia gravis, antibodies, clinical aspects, outcome, epidemiology
Dirección postal: Florencia Aguirre, Hospital Ramos Mejía, Urquiza 609, 1221 Buenos Aires, Argentina
e-mail: aguirreflor@gmail.com
La miastenia gravis (MG) es una enfermedad autoinmune mediada por anticuerpos dirigidos contra proteínas post-sinápticas de la unión neuromuscular. La alteración de la trasmisión neuromuscular secundaria a la acción de estos anticuerpos, determina las manifestaciones clínicas típicas de esta entidad, caracterizadas debilidad fluctuante que involucra combinaciones variables de músculos oculares, bulbares, de extremidades y respiratorios 1-3.
La mayoría de los enfermos presentan anticuerpos dirigidos contra el receptor nicotínico de acetilcolina (ACRA); estos anticuerpos son identificados en el 85% de los que padecen MG generalizada y en alrededor del 50% de los que presentan MG ocular 4, 5. En una proporción variable de los pacientes ACRA negativos, se identifican otros anticuerpos con valor patogénico, como el anticuerpo contra tirosina quinasa músculo específica (anti-MuSK) o el anticuerpo contra la lipoproteína de baja densidad 4 (anti-LRP4)6.
Los datos de incidencia y prevalencia de MG varían ampliamente; la incidencia ha sido comunicada entre 1.7 y 21 casos por millón por año, y su prevalencia en hasta 150 a 250 por millón 7. Un estudio de MG en nuestro país, comunicó una densidad de incidencia ajustada a la población argentina de 38.8/1 000 0008. Esta enfermedad afecta todas las etnias y si bien puede manifestarse a cualquier edad se reconocen dos picos de incidencia, un primer pico en entre los 20 y 40 años con predominio femenino y otro después de los 60 años con mayor representación masculina 7, 9-11.
Cada vez es más reconocido que la MG es una enfermedad heterogénea que incluye diferentes subgrupos de pacientes. Es posible clasificar a la MG en subtipos de acuerdo a su edad de inicio (de inicio temprano vs. de inicio tardío), a la extensión del compromiso clínico (ocular puro vs. generalizado), al anticuerpo responsable (ACRA positivo, anti-MuSK positivo, LRP4 positivo o seronegativa) o a la enfermedad tímica asociada (timomatosa o no timomatosa) 12. El objetivo de este estudio fue describir los aspectos clínicos, epidemiológicos, serológicos y la evolución de pacientes con MG en seguimiento en un Hospital Público de la Ciudad de Buenos Aires.
Materiales y métodos
Se realizó un estudio retrospectivo donde se incluyeron pacientes con diagnóstico de MG asistidos en la Sección de Neuroinmunologia y Electrofisiología del Hospital General de Agudos Dr. José María Ramos Mejía, de la Ciudad Autónoma de Buenos Aires, en el período comprendido entre los años 2008 y 2018.
El diagnóstico de MG se realizó en base a los resultados de parámetros clínicos y de laboratorio convencionales: debilidad muscular fatigable, presencia de anticuerpos ACRA o anti-MuSK, respuesta clínica positiva a inhibidores de acetilcolinesterasa y estudios electrofisiológicos compatibles con afección de la transmisión neuromuscular (estimulación repetitiva o electromiografía de fibra única).
Se revisaron historias clínicas de personas con diagnóstico de MG, de las cuales se recolectaron datos referentes al sexo, edad de inicio de los síntomas y de diagnóstico de la enfermedad, síntoma de presentación, hallazgos tímicos y presencia de enfermedades autoinmunes asociadas. También se consignaron datos referidos al subtipo serológico, la gravedad y evolución clínica de la enfermedad y el tratamiento requerido en cada caso.
Las determinaciones de anticuerpos ACRA y anti-MuSK se realizaron por radioinmunoanálisis; resultados de ACRA mayores a 0.5 nMol/l de ACRA y de anticuerpos anti-MuSK mayores a 0.05 nMol/l, fueron considerados positivos.
La enfermedad tímica fue diagnosticada mediante estudios por imágenes del torax (tomografía computarizada o resonancia magnética). En 44 pacientes que fueron timectomizados, se obtuvieron datos referidos a la anatomía patologica.
El screening de enfermedad tiroidea mediante detección sérica de las hormonas tiroideas T3, T4, TSH y anticuerpos anti peroxidasa se solicitó en forma rutinaria a todos los pacientes.
La búsqueda y diagnóstico de otras enfermedades autoinmunes asociadas fue guiada en función de signos o
síntomas sugestivos en cada caso.
La gravedad clínica de la MG fue evaluada según la escala MGFA (Myasthenia Gravis Foundation of America) 13 en el momento de máxima gravedad en cada caso. También se valoró la evolución clínica determinada por la clasificación post-intervention status (PIS) en la última consulta.
Los datos obtenidos fueron volcados en una base de datos y luego analizados empleando el software estadístico SPSS versión 25. Para los análisis de correlación se utilizó la prueba de correlación de Pearson y para la comparación de medias un t-test de dos colas para muestras no apareadas.
Resultados
Se incluyeron 190 pacientes con diagnóstico de MG, 119 mujeres (62.6%) y 71 hombres. En el total, la relación mujer-hombre (m/h) fue de 1.7/1, aunque esta proporción no fue homogénea en todos los rangos etarios. Las diferencias de género, según la edad de inicio de la MG, se profundizaron hacia los extremos (Fig. 1). En los que comenzaron tempranamente (menores de 16 años) el predominio femenino fue muy marcado con una relación de 12/1. La mayor representación de mujeres se mantuvo en pacientes que comenzaron antes de los 50 años, mientras que a partir de la sexta década de la vida se observó un predominio masculino que se intensifico en aquellos con inicio de enfermedad después de los 60 años, con una relación hombre-mujer de 2.7/1.
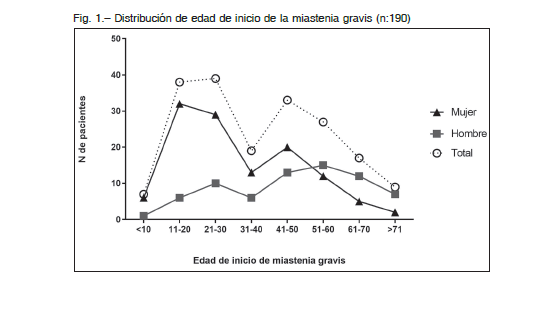
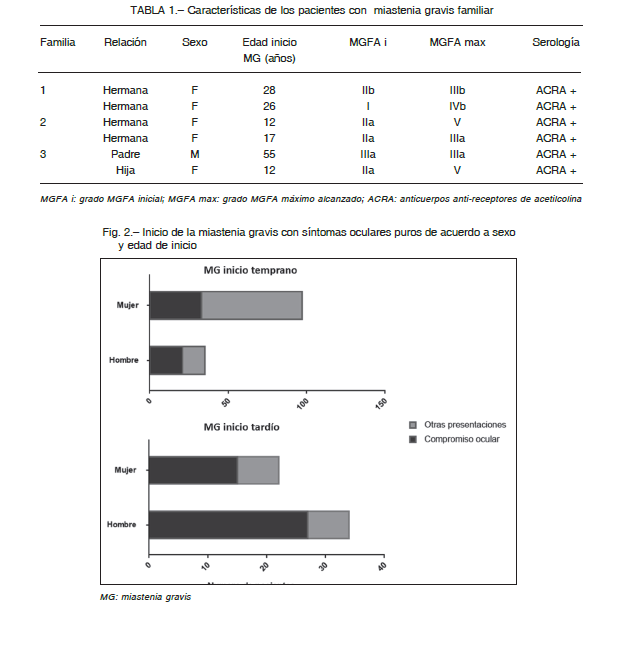
La edad media de inicio de MG fue 38 años (rango 3 a 81 años). El 69.5% (132) fue considerado como MG de inicio temprano (inicio de la enfermedad antes de los 50 años) y el 30.5% (58) como MG de inicio tardío (después de los 50 años). Las mujeres iniciaron la enfermedad más tempranamente, 32 vs. 48 años en los hombres (p < 0.0001). La duración media de la enfermedad fue de 10.6 años (rango 6 meses a 63 años). La MG familiar autoinmune representó el 3.2% de los casos; 6/190 pacientes pertenecientes a familias no relacionadas tenían un familiar de primer grado afectados por la enfermedad (Tabla 1).
El inicio de MG con síntomas oculares exclusivos se observó en el 52% de los pacientes, con debilidad muscular limitada a los miembros en el 22%, con compromiso de músculos bulbares en el 18% y con debilidad muscular generalizada en el 8%.
Del total de 91 pacientes que comenzaron con manifestaciones oculares, 72 (79.1%) desarrollaron síntomas de compromiso generalizado durante el trascurso de la enfermedad. El 84.1% de los que generalizaron, lo hicieron dentro del primer año desde el inicio de la sintomatología.
La proporción de los que iniciaron con síntomas oculares fue diferente entre distintos grupos etarios (Fig. 2).
Esta forma de presentación fue significativamente más frecuente en aquellos de inicio tardío que en los de inicio temprano, 74% vs. 42% respectivamente (p < 0.001) y más en hombres que en mujeres, 70% vs. 41% respectivamente (p < 0.001).
El 12.1% (23) persistieron con síntomas oculares puros mas allá de los dos años de inicio de la enfermedad y fueron considerados MG ocular.
La gravedad de la MG según el máximo grado MGFA alcanzado fue: clase I en 23, clase IIa-IIb en 38, clase IIIa-IIIb en 75, clase IVa-IVb en 28 pacientes y clase V en 27. En dos de los 27 que presentaron al menos una crisis miasténica (7.4%), esta fue la forma de inicio de la enfermedad.
De acuerdo a la clasificación post-intervention status (PIS) evaluado en la última consulta, 19 se encontraban en remisión (7 en remisión crónica estable y 12 en remisión farmacológica), 77 en estado de manifestaciones mínimas, 53 mejoraron, 17 exacerbaron sus síntomas y 13 no presentaron cambios.
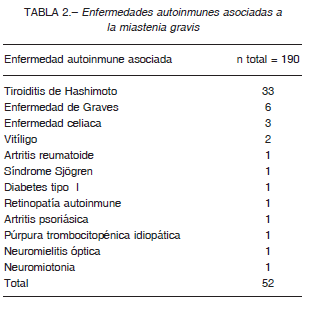
El 27.1% presentó otra enfermedad autoinmune asociada. Los desórdenes tiroideos autoinmunes fueron lo más frecuentes (81.2%), 33 pacientes con tiroiditis de Hashimoto y 6 con enfermedad de Graves (Tabla 2).
Se realizó una evaluación rutinaria de enfermedad tímica con estudios por imagen torácico (resonancia magnética nuclear o tomografía axial computarizada) en 165 pacientes. Se observó una imagen en mediastino anterior compatible con timoma en el 11.5% (19/165). Del resto de los pacientes, 30 presentaron hallazgos compatibles con hiperplasia tímica y en 116 no se observaron anormalidades a nivel del mediastino anterior.
Entre los pacientes con MG timomatosa la media de edad de inicio fue de 47 años, que fue significativamente más tardía que en aquellos sin timoma, 39 años (p <0.005). La evolución de la MG fue más tórpida, con mayor frecuencia de crisis miasténicas en el seguimiento, 31.6% vs. 10.3% (p < 0.05).
El diagnóstico de timoma fue realizado en screening torácico inicial a partir del diagnostico de MG en el 63.2% de los casos. Del resto de los pacientes, 5 desarrollaron MG posteriormente al diagnóstico de timoma, con una media de 4 años (rango 2 meses a 13 años) y en dos casos el timoma fue detectado años después del diagnóstico de MG (5 años y 8 años después). El estudio por imágenes de torax inicial era normal en estos 2 pacientes.
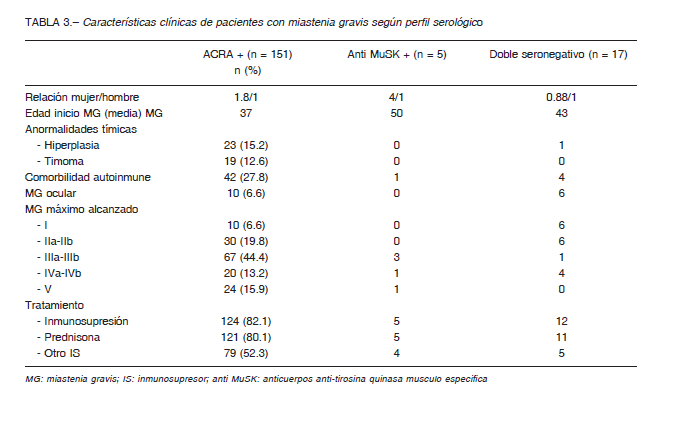
Una determinación de ACRA fue realizada en 183/190, de estos, 151 resultaron positivos (82.5%). La proporción de ACRA positivos fue de 84.2% en MG generalizada y de 52.6% en MG ocular (p < 0.05). No hubo diferencias estadísticamente significativas en cuanto a ACRA seropositividad según género ni edad de inicio.
De 34 ACRA negativos, se solicitó anti-MuSK en 22 pacientes; se diagnosticó MG anti-MuSk positiva en 5/22
(22.7%) de ellos. De 171 testeados para ACRA y MuSK, 5 (2.9%) fueron anti-Musk positivos; estos mostraron un predominio femenino marcado (80%) y una media de edad de inicio de MG más tardía (49.6 años) (Tabla 3).
La gran mayoría requirió inmunosupresión para control de la sintomatología de la enfermedad. El 78% recibió corticoides durante algún período de la enfermedad y cerca de la mitad (48%) requirió el uso de un inmunosupresor no esteroideo, más frecuéntenme azatioprina. La instauración de tratamiento esteroideo se realizó con dosis bajas iniciales que fueron incrementadas progresivamente hasta recibir dosis de 1 mg/kg/día o hasta alcanzar estado de manifestaciones mínimas de la enfermedad. La dosis máxima media de predniosona administrada fue de 36 mg/día.
Por otro lado, 37/190 pacientes, el 19.5 %, fue controlado exitosamente solo con tratamiento sintomático (la dosis promedio de piridostigmina fue de 336 mg/día); la media de duración de MG en este grupo fue de 9.8 años, similar a los que fueron inmunosuprimidos que fue de 9.0 años.
Discusión
Los aspectos demográficos hallados en nuestra población de pacientes con MG permiten reproducir la distribución bimodal previamente comunicada 7,9-10. En el análisis de nuestra serie, es posible diferenciar un primer pico de incidencia en menores de 40 años, con un franco predominio femenino y un segundo pico en mayores de 40 años con mayor representación masculina que se profundiza después de los 60 años. Este diferente predominio de género de acuerdo a la edad del debut de la MG se vuelve evidente al comparar las medias de edad al diagnóstico, resultando significativamente más avanzada en hombres que en mujeres. Si bien esta característica fue previamente informada en pacientes con MG, así como en otras enfermedades autoinmunes, esta diferencia de más de 15 años de edad media de inicio
entre ambos sexos, fue más marcada que lo descripto previamente 11, 14. Factores hormonales probablemente se relacionen con la diferente incidencia de MG según el sexo. La testosterona juega un rol en la regulación de la respuesta inmune inhibiendo la producción de citoquinas proinflamatorias, la diferenciación TH1, la producción de inmunoglobulinas y aumentando la expresión de citoquinas anti-inflamatorias. La disminución de los niveles de testosterona con la edad justificaría el aumento de la prevalencia de la MG en hombres de edad avanzada. El papel de los estrógenos en la patogénesis de la MG es conocido y se fundamenta en un aumento en la expresión del receptor de estrógenos (ERα) en timocitos y de ERα y ERβ en células T circulantes 15.
La media de edad de inicio fue similar a lo ya comunicado en diferentes series con gran número de pacientes 11, 16-18, sin embargo, fue más temprana que la descripta por otros autores 8, 19.
La MG autoinmune familiar es poco frecuente. Fue posible identificar 6 casos de MG familiar pertenecientes a 3 familias no relacionadas, constituyendo una frecuencia de 3.2% de MG familiar, similar a lo descripto en una población española 14.
Más de la mitad se presentó con síntomas oculares puros. El compromiso ocular como forma de presentación más frecuente fue ampliamente descripto en otras series de pacientes con MG 15-20. Sin embargo, de igual manera que en otras descripciones, la gran mayoría desarrolló síntomas generalizados en el trascurso de la enfermedad 23, 24.
Si bien la forma de presentación con compromiso ocular fue la más frecuente en todas las edades, nosotros encontramos una diferencia marcada en la representación de este síntoma en distintos subgrupos de pacientes, hallándola muy prevalente en hombres con inicio tardío.
En este subgrupo, más del 80% comenzó con síntomas oculares, en contraste con solo un tercio de las mujeres de inicio temprano. Luego de dos años del inicio de la enfermedad, el porcentaje de pacientes considerados como MG ocular fue del 12.1%, similar a lo previamente comunicado 11, 25.
La gran mayoría presentó una evolución favorable, el 78.4% había mejorado hacia la última evaluación, y de estos el 65% se encontraba en estado de remisión o manifestaciones mínimas. Estos datos son similares a los informados por Tsinzerling y col. 26. El 14% presentó al menos una crisis miasténica, proporción que se encuentra dentro de lo esperado según lo publicado previamente27-29.
Se halló al menos una enfermedad autoinmune asociada a la MG en cerca de un tercio de los pacientes de los casos; esta proporción es más elevada que lo publicado previamente por otros autores 16,30 y representa más del doble de lo estimado en una revisión sistemática de trabajos observacionales que informaron enfermedades autoinmunes asociadas a la MG 31. De igual forma que lo descripto previamente, los desórdenes tiroideos autoinmunes fueron los más frecuentes 32.
En la mayoría de los casos, las imágenes de tórax no revelaron anormalidades tímicas. Solo hubo hallazgos compatibles con hiperplasia tímica en el 18.2% y con timoma en el 11.5% de los casos. Estas proporciones son muy similares a las comunicadas por Karni y col. 33; sin embargo, la frecuencia de hiperplasia tímica fue menor a la descripta por otros autores 34, 35. La relación entre enfermedad tímica y la gravedad de la MG fue valorada por otros autores con resultados disímiles 36-39. En nuestra serie, los pacientes con MG asociada a timoma se caracterizaron por presentar una mayor gravedad de la MG, un tercio de ellos tuvo al menos una crisis miasténica en la evolución.
Desde el punto de vista serológico más del 80% de nuestra población fue MG ACRA positiva. MG anti-MuSK positiva represento menos del 3% y MG doble seronegativa casi el 10%. La proporción de pacientes anti-MuSK positivos varía en las distintas series según su localización geográfica; esta situación probablemente se relaciona con factores genéticos, ambientales o ambos. Hubo valores más altos de prevalencia en regiones cercanas al Ecuador y áreas de más baja prevalencia hacia los polos 40, 41. A pesar de las diferencias étnicas, la proporción de MG anti-MuSK de 2.93% observada en nuestra serie, es similar a la observada en una población australiana (3.4%) con una latitud similar a la de la Argentina 42.
Con respecto al tratamiento recibido, si bien la mayoría de los pacientes requirieron inmunosupresión en algún momento de la evolución, se destaca un subgrupo de alrededor del 20% del total que fueron exitosamente medicados con piridostigmina como único tratamiento.
Esta experiencia es similar a la de otros autores 17, 43-47. Nuestro estudio comunica las características clínicas, demográficas y serológicas de una serie de pacientes con MG en seguimiento en un centro único de la ciudad de Buenos Aires en un período de 10 años.
Los datos obtenidos en los enfermos de nuestra población son similares en su mayoría, a lo previamente descripto por otros autores. Sin embargo, la diferente predominancia de géneros en relación a la edad de inicio de la MG, que se profundiza más marcadamente hacia los extremos de la vida, el inicio con compromiso ocular más frecuente en hombres con inicio tardío de MG, y una mayor frecuencia de comorbilidad autoinmune en nuestra serie, podrían ser características propias de nuestra población. Futuros estudios locales permitirán aclarar estos aspectos.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Gilhus NE. Myasthenia and neuromuscular junction. Curr Opin Neurol 2012; 25: 523-9.
2. Querol L, Illa I. Myasthenia and the neuromuscular junction. Curr Opin Neurol 2013; 26: 459-65.
3. Verschuuren JJ, Huijbers MG, Plomp JJ, et al. Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase, and low-density lipoprotein receptor-related protein 4. Autoimmune Rev 2013; 12: 918-23.
4. Lennon VA, Lindstrom JM, Seybold ME. Experimental autoimmune myasthenia: a model of myasthenia gravis
in rats and guinea pigs. J Exp Med 1975; 141: 1365-75.
5. Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology 1976; 26: 1054-9.
6. Zisimopoulou P, Brenner T, Trakas N, Tzartos SJ. Serological diagnostics in myasthenia gravis based on novel assays and recently identified antigens. Autoimmun Rev 2013; 12: 924-30.
7. Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol 2010; 10:46.
8. Bettini M, Chaves M, Cristiano E, et al. Incidence of autoimmune myasthenia gravis in a health maintenance organization in Buenos Aires, Argentina. Neuroepidemiology 2017; 48: 119-23.
9. Heldal AT, Owe JF, Gilhus NE, Romi F. Seropositive myasthenia gravis; a nationwide epidemiologic study. Neurology 2009; 73: 150-1.
10. Andersen JB, Engeland A, Owe JF, Gilhus NE. Myasthenia gravis requiring pyridostigmine treatment in a national population cohort. Eur J Neurol 2010; 17: 1445-50.
11. Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle Nerve 2008; 37: 141-9.
12. Romi F, Hong Y, Gilhus NE. Pathophysiology and immunological profile of myasthenia gravis and its subgroups. Curr Opin Immunol 2017; 49: 9-13.
13. Jaretzki A 3rd, Barohn RJ, Ernstoff RM, Kaminski HJ, Keesey JC, Penn AS, Sanders DB. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Ann Thorac Surg 2000;70:327-34.
14. Salvado M, Canela M, Ponseti JM, et al. Study of the prevalence of familial autoimmune myasthenia gravis in a Spanish cohort. J Neurol Sci 2016; 360: 110-4.
15. Asmail A, Kesler A, Kolb H, Drory VE, Karni A. A tri-modal distribution of age-of-onset in female patients with myasthenia gravis is associated with the gender-related clinical differences. Int J Neurosci 2019; 129: 313-9.
16. Giagheddu M, Puggioni G, Sanna G, et al. Epidemiological study of myasthenia gravis in Sardinia, Italy (1958-1986). Acta Neurol Scand 1989; 79: 326-33.
17. Aguiar Ade A, Carvalho AF, Costa CM, et al. Myasthenia gravis in Ceará, Brazil: clinical and epidemiological aspects. Arq Neuropsiquiatr 2010; 68: 843-8
18. Oöpik M1, Kaasik AE, Jakobsen J. A population based epidemiological study on myasthenia gravis in Estonia. J Neurol Neurosurg Psychiatry 2003; 74: 1638-43.
19. Sanders DB, Evoli A. Immunosuppressive therapies in myasthenia gravis. Autoimmunity 2010; 43: 428-35.
20. Robertson NP, Deans J, Compston DA. Myasthenia gravis: a population based epidemiological study in Cambridgeshire, England. J Neurol Neurosurg Psychiatry 1998; 65: 492-6.
21. Zhang X, Yang M, Xu J, et al. Clinical and serological study of myasthenia gravis in HuBei Province, China. J Neurol Neurosurg Psychiatry 2007; 78: 386-90.
22. Oosterhuis HJ. The natural course of myasthenia gravis: a long term follow up study. J Neurol Neurosurg Psychiatry 1989; 52: 1121-7.
23. Bever CT Jr, Aquino AV, Penn AS, Lovelace RE, Rowland LP. Prognosis of ocular myasthenia. Ann Neurol 1983; 14: 516-9.
24. Aguirre F, Villa AM. Prognosis of ocular myasthenia gravis in an Argentinian population. Eur Neurol 2018; 79: 113-7.
25. Gilhus, NE. Myasthenia Gravis. N Eng J Med 2016; 375: 2570-81.
26. Tsinzerling N, Lefvert AK, Matell G, Pirskanen-Matell R. Myasthenia gravis: a long term follow-up study of Swedish patients with specific reference to thymic histology. J Neurol Neurosurg Psychiatry 2007; 78: 1109-12.
27. Matsui N, Nakane S, Nakagawa Y, et al. Increasing incidence of elderly onset patients with myasthenia gravis in a local area of Japan. J Neurol Neurosurg Psychiatry 2009; 80: 1168-71.
28. Jani-Acsadi A, Lisak RP. Myasthenic crisis: Guidelines for prevention and treatment. J Neurol Sci 2007; 261: 127-33.
29. Hehir MK, Silvestri NJ. Generalized myasthenia gravis: classification, clinical presentation, natural istory, and epidemiology. Neurol Clin 2018; 36: 253-60.
30. Poulas K, Tsibri E, Kokla A, et al. Epidemiology of seropositive myasthenia gravis in Greece. J Neurol Neurosurg Psychiatry 2001; 71: 352-6.
31. Mao ZF, Yang LX, Mo XA, et al. Frequency of autoimmune diseases in myasthenia gravis: a systematic review. Int J Neurosci 2011; 121: 121-9.
32. Lopomo A, Berrih-Aknin S. Autoimmune thyroiditis and myasthenia gravis. Front Endocrinol (Lausanne) 2017; 8: 169.
33. Karni A, Asmail A, Drory VE, Kolb H, Kesler A. Thymus involvement in myasthenia gravis: Epidemiological and clinical impacts of different self-tolerance breakdown mechanisms. J Neuroimmunol 2016; 298:58-62.
34. Levinson AI, Song D, Gaulton G, Zheng Y. The intrathymic pathogenesis of myasthenia gravis. Clin Dev Immunol 2004; 11:215-20.
35. Tsinzerling N, Lefvert AK, Matell G, Pirskanen-Matell R. Myasthenia gravis: a long term follow-up study of Swedish patients with specific reference to thymic histology. J Neurol Neurosurg Psychiatry 2007; 78: 1109-12.
36. Roncoroni AJ, Herrera MR, Rosenberg M. Timectomía en miastenia gravis. Medicina (B Aires) 1978; 38: 9-19.
37. Liu W, Tong T, Ji Z, Zhang Z. Long-term prognostic analysis of thymectomized patients with myasthenia gravis. Chin Med J (Engl) 2002; 115: 235-7.
38. Romi F, Gilhus NE, Aarli JA. Myasthenia gravis: clinical, immunological, and therapeutic advances. Acta Neurol Scand 2005; 111: 134-4.
39. Romi F, Gilhus NE, Varhaug JE, Myking A, Aarli JA. Disease severity and outcome in thymoma myasthenia
gravis: a long-term observation study. Eur J Neurol 2003; 10: 701-6.
40. Romi F, Gilhus NE, Aarli JA. Myasthenia gravis: disease severity and prognosis. Acta Neurol Scand Suppl 2006; 183: 24-5.
41. Deymeer F, Gungor-Tuncer O, Yilmaz V, et al. Clinical comparison of anti-MuSK- vs anti-AChR-positive and seronegative myasthenia gravis. Neurology 2007; 68: 609-11.
42. Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 2011; 44: 36-40.
43. Blum S, Lee D, Gillis D, McEniery D, Reddel S, McCombe P. Clinical features and impact of myasthenia gravis disease in Australian patients. J Clin Neurosci 2015; 22: 1164-9.
44. Beekman R, Kuks JB, Oosterhuis HJ. Myasthenia gravis: diagnosis and follow-up of 100 consecutive patients. J Neurol 1997; 244: 112-8.
45. Mantegazza R, Beghi E, Pareyson D, et al. A multicenter follow-up study of 1152 patients with myasthenia gravis in Italy. J Neurol 1990; 237: 339-44.
46. Cea G, Martinez D, Salinas R, Vidal C, Hoffmeister L, Stuardo A. Clinical and epidemiological features of myasthenia gravis in Chilean population. Acta Neurol Scand 2018; 138: 338-43.
47. Rey RD, Buzzi AE, Astudillo MA, et al. Diagnóstico y tratamiento de la myasthenia gravis. Experiencia 1974-1992. Medicina (B Aires) 1995; 55: 11-20.
– – – –
In the practical use of our intellect, forgetting is as important as remembering
En el uso práctico de nuestro intelecto, olvidar es tan importante como recordar
William James (1842-1910)
The principles of Psychology, 1890