
M. SILVINA ODSTRCIL-BOBILLO 1, 2, DANA KOHAN 3, PAULA G. HELLER 4, 5, VICTORIA OTERO 1, 2, MARÍA PAULA RUSSO 1, ANA L. BASQUIERA 1, 2
1Departamento de Medicina Interna, Hospital Italiano de Buenos Aires, 2 Servicio de Hematología, Hospital Italiano de Buenos Aires, 3 Servicio Anatomía Patológica, Hospital Italiano de Buenos Aires, 4 Servicio Hematología Investigación, Instituto de Investigaciones Médicas Alfredo Lanari, Universidad de Buenos Aires, Argentina, 5 Departamento Hematología Investigación, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Universidad de Buenos Aires (UBA), Instituto de Investigaciones Médicas (IDIM), Buenos Aires, Argentina
Resumen El síndrome de Wiskott-Aldrich (SWA) es un raro síndrome de inmunodeficiencia primaria ligado al cromosoma X que se asocia con aumento de incidencia de infecciones, trastornos autoinmunes y neoplasias. Se presenta el caso de un varón de 41 años con diagnóstico de síndrome de Wiskott-Aldrich y cuadro de ileítis como forma de presentación de un síndrome linfoproliferativo. La ileítis, en el contexto del paciente, representa un problema clínico dado el gran número de diagnósticos diferenciales (enfermedad inflamatoria intestinal, infecciones, neoplasias y enfermedades linfoproliferativas) por lo que suele requerir diagnóstico anatomopatológico y consideraciones particulares respecto al posterior tratamiento específico.
Palabras clave: Wiskott-Aldrich, ileítis, síndrome linfoproliferativo
Abstract Wiskott-Aldrich syndrome is a rare X chromosome-linked primary immunodeficiency syndrome associated with an increased incidence of infections, autoimmune disorders and neoplasms. We present the case of a 41-year-old man with a diagnosis of Wiskott-Aldrich syndrome with ileitis as a form of presentation of a lymphoproliferative syndrome. The ileitis, in the context of the patient, represents a clinical challenge given the large number of differential diagnoses (inflammatory bowel disease, infections, neoplasms and lymphoproliferative diseases), so it usually requires anatomopathological diagnosis and particular considerations regarding the subsequent specific treatment.
Key words: Wiskott-Aldrich, ileitis, lymphoproliferative syndrome
Dirección postal: María Silvina Odstrcil Bobillo, Hospital Italiano Buenos Aires, Tte. General J. D. Perón 4190, 1199 Buenos Aires, Argentina
e-mail: maria.odstrcil@hospitalitaliano.org.ar
El síndrome de Wiskott-Aldrich (SWA) es una rara inmunodeficiencia primaria con una prevalencia de 1/1 000 000 habitantes, causada por una mutación del gen de la proteína del SWA, que se encuentra expresado únicamente por células hematopoyéticas 1.
La proteína del SWA tiene un rol fundamental en la inmunidad innata y adquirida a través de la regulación de procesos celulares dependientes del citoesqueleto de actina incluyendo la sinapsis inmune, señalización celular, migración y liberación de citoquinas 2. Características más complejas de SWA, como la asociación a fenómenos autoinmunes e inflamación, provienen de la identificación de defectos en el número y función de las células mieloides, dendríticas y las células T y B reguladoras, así como defectos en las interacciones positivas (centro germinal del nodo) y negativas (autorreactividad) de células B 3.
Los trastornos gastrointestinales (diarrea, malabsorción, dolor abdominal) están presentes entre 5 y 50% de sujetos con inmunodeficiencias primarias, y pueden ser de etiología inflamatoria, infecciosa, autoinmune o neoplásica 4. La prevalencia en pacientes con SWA de enfermedad inflamatoria intestinal semejante a la colitis ulcerosa está informada en un 10% 5.
Muchos de los trastornos gastrointestinales imitan las formas clásicas de alguna enfermedad (en ausencia de inmunodeficiencia) como la celíaca o la enfermedad inflamatoria intestinal, pero difieren en la patogenia y a menudo no responden a las terapias convencionales, por lo que su diagnóstico es difícil y suele requerir estudios histológicos 6
Caso clínico
Varón de 41 años con antecedente de trombocitopenia ligada al X por mutación del gen SWA (mutación p.Gln52His en el exón 2 del gen SWA) diagnosticado en la infancia, en seguimiento por lesiones cutáneas de tipo eczematosas y trombocitopenia crónica con microplaquetas (con recuento de plaquetas habitual de 30 × 109/l), que años previos a la consulta realizó tratamiento con eltrombopag, gammaglobulina y soporte transfusional por hematoma extradural espontáneo.
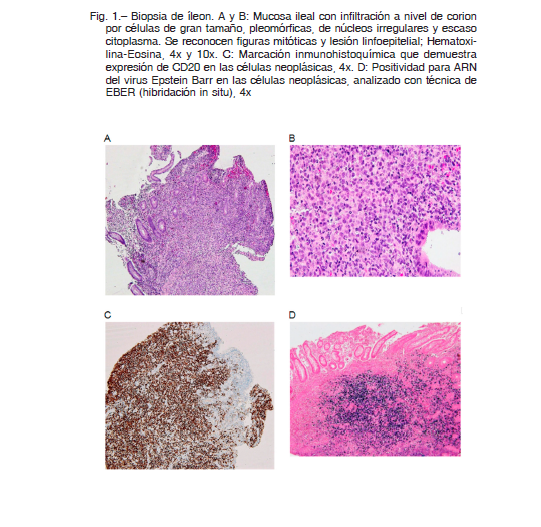
Consultó por dolor abdominal en fosa ilíaca derecha de un mes de evolución sin otra sintomatología asociada; como datos relevantes del laboratorio se observó: plaquetopenia (20 × 109/l), eritrosedimentación elevada (40 mm) y serologías para HIV, citomegalovirus, anticuerpos anti músculo liso y anticuerpos anti-Saccharomyces cerevisiae negativas. La ecografía abdominal mostró un proceso inflamatorio inespecífico a nivel del íleon por lo que se inició tratamiento con ciprofloxacina. Se realizó tomografía computarizada de abdomen donde se evidenció engrosamiento parietal del íleon terminal asociado a aumento en la densidad de planos adiposos vecinos y adenomegalias regionales. Se completó 14 días de ciprofloxacina sin mejoría clínica y, por persistencia de lesiones en la tomografía abdominal se realizó videocolonoscopia, en la que se observó a nivel de íleon terminal mucosa polinodular, congestión y ulceración con fibrina que comprometía el 50% de la circunferencia. La anatomía patológica de la biopsia fue compatible con linfoma difuso de células grandes B, fenotipo centro germinal (Fig. 1A, B, C, D). Se realizaron técnicas de inmunohistoquímica, CD3: negativo, CD20: positivo, CD30: positivo, EBER positivo, Ki-67: índice proliferativo del 60%, BCL6: positivo. Se completó la evaluación con tomografía por emisión de positrones, que informó aumento de metabolismo a nivel del íleon terminal (SUV 27) y conglomerado en raíz del mesenterio adyacente, asociado a hipercaptación difusa en la médula ósea sin evidencia de lesiones focales. Se concluyó estadio 1 A, extranodal, IPI 0.
El paciente completó cuatro ciclos de quimioterapia con esquema R-CHOP con dosis ajustadas de doxorrubicina al 75% por plaquetopenia, presentando remisión completa. Posteriormente se realizó trasplante alogénico de células progenitoras hematopoyéticas con donante no relacionado 10 de 10, utilizando acondicionamiento de intensidad reducida, logrando recuperación hematológica completa; presentó injerto de plaquetas al día +15 postrasplante con volumen plaquetario normal y valores normales desde el día +29. Como complicaciones postrasplante presentó enfermedad injerto contra huésped cutáneo grado II con requerimiento de tratamiento esteroideo y cistitis por virus BK.
En controles postrasplante se constató quimerismo del donante mayor del 95%, y disminución del grosor y metabolismo de la lesión intestinal del íleon terminal, con aumento difuso de fijación (SUV 11.9; previo 27), asociado a persistencia de ganglios en mesenterio sin concentración el radiotrazador. Se realizó biopsia, que informó hiperplasia folicular, y se inició tratamiento con budesonide con buena respuesta. Actualmente, al año del trasplante, se encuentra sin recaída de la enfermedad.
Discusión
La presentación del cuadro de ileítis en un paciente con SWA es un problema diagnóstico dado la presentación particular de los síntomas y el número de etiologías posibles (enfermedad inflamatoria intestinal, infecciones, neoplasias y enfermedades linfoproliferativas).
Aproximadamente el 18% de los pacientes con SWA desarrollarán algún tipo de malignidad, con una edad promedio de inicio de 9.5 años. Esto corresponde a un riesgo relativo de malignidad 100 veces mayor al de la población general 7.
El linfoma no Hodgkin (LNH) representa el 60% de los tumores asociados a trastornos inmunes primarios y el 75% en pacientes con SWA, siendo el más frecuente en esta enfermedad 8.
El LNH asociado al SWA tiende a ser más agresivo, diseminado al momento del diagnóstico y a presentarse en tejidos extraganglionares, siendo el cerebro el órgano más comúnmente afectado, seguido de la piel, intestino delgado, hígado y pulmón 9. Presentan además un peor pronóstico que en individuos sin SWA, con una supervivencia menor a 5% en 2 años, aunque hay comunicaciones que informan mayor supervivencia 10. El tipo histológico más común es linfoma difuso de células B grandes, similar a lo encontrado en trastornos linfoproliferativos post trasplante o Sida 11.
Cabe destacar que síndromes linfoproliferativos asociados a virus de Epstein Barr (VEB) son más frecuentes en pacientes con inmunodeficiencias como el SWA. El mecanismo de esta asociación puede relacionarse con una respuesta anormal de los linfocitos a la infección por VEB secundaria a la expresión superficial reducida y la proteólisis aberrante de CD23 en SWA 12.
El tratamiento del linfoma en SWA es difícil ya que el estado de inmunodeficiencia previo trae aparejado mayores tasas de complicaciones infecciosas y mielotoxicidad, por lo que se suele requerir ajustes de dosis, como en el caso presentado 13. Los esquemas quimioterápicos que incluyen el anticuerpo anti-CD20 se asocian a mejores resultados 14.
El trasplante alogénico de células progenitoras hematopoyéticas con un donante compatible es la única terapia curativa disponible para pacientes con SWA 15.
La importancia de la presentación de este caso radica en que se trata de una enfermedad rara con una complicación que si bien es típica, presenta múltiples diagnósticos diferenciales y una atención particular y personalizada, que comprende tanto al internista como al hematólogo.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Rivers E, Thrasher AJ. Wiskott-Aldrich syndrome protein: emerging mechanisms in immunity. Eur J Immunol 2017; 47: 1857-66.
2. Recher M, Burns SO, de la Fuente MA, et al. B cell-intrinsic deficiency of the Wiskott-Aldrich syndrome protein (WASp) causes severe abnormalities of the peripheral B-cell compartment in mice. Blood 2012; 119: 2819-28.
3. Castiello MC, Bosticardo M, Pala F, et al. Wiskott-Aldrich Syndrome protein deficiency perturbs the homeostasis of B-cell compartment in humans. J Autoimmun 2014; 50: 42-50.
4. Agarwal S, Mayer L. Diagnosis and treatment of gastrointestinal disorders in patients with primary immunodeficiency. Clin Gastroenterol Hepatol 2013; 11: 1050–63.
5. Folwaczny C, Ruelfs C, Walther J, König A, Emmerich B. Ulcerative colitis in a patient with Wiskott-Aldrich syndrome. Endoscopy 2002; 34: 840-1.
6. Dupuis-Girod S, Medioni J, Haddad E, et al. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics 2003; 111: 622-7.
7. Mueller BU, Pizzo PA. Cancer in children with primary or secondary immunodeficiencies. J Pediatr 1995; 126: 1-10.
8. Du S, Scuderi R, Malicki DM, et al. Hodgkin’s and non- Hodgkin’s lymphomas occurring in two brothers with Wiskott-Aldrich syndrome and review of the literature. Pediatr Dev Pathol 2011; 14: 64-70.
9. Cotelingam JD, Witebsky FG, Hsu SM, Blaese RM, Jaffe ES. Malignant lymphoma in patients with the Wiskott- Aldrich syndrome. Cancer Invest 1985; 3: 515-22.
10. Bosticardo M, Marangoni F, Aiuti A, Villa A, Grazia Roncarolo M. Recent advances in understanding the
pathophysiology of Wiskott-Aldrich syndrome. Blood 2009; 113: 6288-95.
11. Sebire NJ, Haselden S, Malone M, Davies EG, Ramsay AD. Isolated EBV lymphoproliferative disease in a child with Wiskott-Aldrich syndrome manifesting as cutaneous lymphomatoid granulomatosis and responsive to anti-CD20 immunotherapy. J Clin Pathol 2003; 56: 555-7.
12. Simon HU, Higgins EA, Demetriou M, et al. Defective expression of CD23 and autocrine growth-stimulation in Epstein-Barr virus (EBV)-transformed B cells from patients with Wiskott-Aldrich syndrome (WAS). Clin Exp Immunol 1993; 91: 43-9.
13. Sebire NJ. Isolated EBV lymphoproliferative disease in a child with Wiskott-Aldrich syndrome manifesting as cutaneous lymphomatoid granulomatosis and responsive to anti-CD20 immunotherapy. J Clin Pathol 2003; 56: 555–7.
14. Senapati J, Devasia AJ, David S, et al. Diffuse large B cell lymphoma in wiskott-Aldrich syndrome: a case report and review of literature. Indian J Hematol Blood Transfus 2014; 30: 309-13.
15. Filipovich AH, Stone JV, Tomany SC, et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry and the National Marrow Donor Program. Blood 2001; 97: 1598-603.