
MARIA CELESTE BUOMPADRE
Servicio de Neurología, Hospital de Pediatría Prof. Dr. Juan P. Garrahan, Buenos Aires, Argentina
Resumen Alrededor del 15% de las epilepsias en pediatría son fármaco-resistentes y en el 40% de este grupo la etiología es una malformación del desarrollo cortical (MDC). El esquema de clasificación actual de las MDC se basa en las etapas primarias de desarrollo de la proliferación celular, migración neuronal y organización cortical. Teniendo en cuenta la clínica y las alteraciones moleculares, se propuso una clasificación basada en la disrupción de las vías principales y el fenotipo neurorradiológico. Se dividió a las MDC en cuatro grupos: la megalencefalia y las displasias corticales focales; las tubulinopatías y lisencefalias; el espectro de las polimicrogirias y las heterotopías. Hasta el momento, más de 100 genes han sido asociados con uno o más tipos de MDC. Los mecanismos biológicos y genéticos incluyen la regulación del ciclo celular en varios estadios, división celular), apoptosis, diferenciación celular, función y estructura del citoesqueleto, migración neuronal y membrana basal. El espectro de síndromes epilépticos asociados con las MDC es amplio e incluye desde encefalopatías epilépticas de comienzo temprano a epilepsias focales de debut más tardío. Teniendo en cuenta que la evolución de la epilepsia hacia la refractariedad en las MDC es importante, el diagnóstico precoz y la elección de la mejor opción terapéutica influirán en el pronóstico de los pacientes.
Palabras clave: malformación cortical, epilepsia, displasia, lisencefalia, tubulinopatías, polimicrogiria
Abstract Malformations of cortical development and epilepsy. Around 15% of childhood epilepsies are resistant to antiepileptic drugs, 40% of which are caused by malformations of cortical development (MCD). The current classification scheme for MCD is based on the primary developmental steps of cell proliferation, neuronal migration, and cortical organization. Considering the clinic and molecular alterations, a classification based on main pathways disruption and imaging phenotype has been proposed. MCD were divided into four groups: megalencephaly and focal cerebral dysplasia; tubulinopathies and lissencephalies; polymicrogyria syndromes and heterotopia syndromes. More than 100 genes have been reported to be associated with different types of MCD. Genetic and biological mechanisms include different stages of cell cycle regulation – especially cell division -, apoptosis, cell-fate specification, cytoskeletal structure and function, neuronal migration, and basement-membrane function. The associated epileptic syndromes are varied ranging from early-onset epileptic encephalopathies to focal epilepsies. As MCD are common causes of refractory epilepsy, a prompt diagnosis and the development of different therapeutic options in order to improve the outcome of the patients are essential.
Key words: cortical malformation, epilepsy, dysplasia, lissencephalies, tubulinopathies, polymicrogyria
e-mail: mariabuompadre@gmail.com
Alrededor del 15% de las epilepsias en pediatría son fármaco-resistentes y en el 40% secundaria a una malformación del desarrollo cortical (MDC). Aproximadamente el 25% de los pacientes con displasia cortical focal evolucionará a epilepsia refractaria 1. Además de los avances en técnicas de resonancia magnética (RM), la identificación de genes y procesos moleculares permitió clarificar estos desórdenes; por ejemplo, las mutaciones pueden tener un efecto variable no solo en el patrón de MDC sino también en su localización 2. Las MDC conducen a la preservación y a la disposición anómala de un número sustancial de células tipo glía. Esta inmadurez en redes neuronales y la actividad del GABA provocan acciones despolarizantes 3. Más de 100 genes han sido descritos hasta el momento en relación a MDC 4.
Clasificación clínica
La clasificación de las MDC se basa en las etapas primarias de desarrollo (proliferación celular, migración neuronal y organización cortical) 5. Si bien el esquema se basa en que el proceso está primariamente afectado, es claro que los desórdenes tempranos influirán en las otras etapas. Además, los genes relacionados con MDC participan en muchos de los estadios del desarrollo cerebral, reforzando el concepto de continuidad (Tabla 1).
Se definen tres grupos: malformaciones secundarias a proliferación anormal neuronal y glial o apoptosis;
malformaciones relacionadas con la migración neuronal anormal y aquellas ligadas al desarrollo migracional y postmigracional. Guerrini y col. 6 propusieron otro esquema basado en disrupción de las vías moleculares principales y fenotipo neurorradiológico. Se definieron cuatro grupos: la megalencefalia y la displasia cortical focal (DCF); las tubulinopatías (TUB) y lisencefalias (LIS); el espectro de las polimicrogirias (PMG) y las heterotopías.
Megalencefalia, hemimegalencefalia y displasias corticales focales
Numerosos genes han sido asociados con PMG y hemimegalencefalia (HMG), especialmente en fenotipos
más graves. La megalencefalia con PMG ocurre en mutaciones del PIK3R2 o de la kinasa serina-treonina (AKT). La HMG aislada más frecuente se ha asociado con mutaciones mosaicas del PIK3CA, AKT3 y de las
vías del MTOR 7. Hay asociaciones con neurofibromatosis tipo 1 (microdeleciones del NF1) que también involucran el gen RNF135, el síndrome de Sotos (con mutaciones del NSD1), y el síndrome de Weaver (mutaciones del EZH2) y también con síndrome de nevus sebáceo linear, esclerosis tuberosa, hemihipertrofia e hipomelanosis de Ito4. Si bien la etiología de las DCF es aún desconocida, en las DCF tipo IIb al igual que en la HMG y los gangliogliomas se ha demostrado un incremento de la ruta de señalización mTOR 8. Las DCF se dividen en 3 tipos y 9 subtipos. La DCF tipo I se caracteriza por laminación cortical anormal, el tipo 2 incluye dislaminación cortical con neuronas dismórficas (2a) y células balonadas (2b) y el tipo 3 ocurre en combinación con otras lesiones cerebrales (tumores) 4. Los hallazgos neurorradiológicos en la HMG son engrosamiento del hemisferio o de parte de uno, pobre diferenciación de la sustancia gris-blanca, señales anormales hipo e hiperintensas en secuencia de T2 en la sustancia blanca y ventriculomegalia homolateral.
Los pacientes con DCF tienen giros lisos y engrosados con incremento de la intensidad de la señal
subcortical en la neuroimagen. En la HMG, la epilepsia es de comienzo temprano y refractaria a fármacos. Las crisis son variables y los espasmos epilépticos son los más frecuentes. Lo que se observa más frecuentemente en el electroencefalograma (EEG) interictal son paroxismo-supresión asimétrico, hipsarritmia asimétrica con actividad de tipo alfa asimétrica y ondas trifásicas asociadas 9.
En la DCF las crisis epilépticas comienzan en la primera década de la vida, generalmente después de los 2 a 3 años, aunque pueden aparecer en el periodo neonatal.
Los pacientes presentan examen neurológico normal y crisis focales motoras complejas o secundariamente
generalizadas. El 50% de los casos puede desarrollar un estado de mal epiléptico al igual que una epilepsia parcial continua. El EEG interictal muestra una actividad de espigas o poliespigas focales de alta frecuencia de descarga que parecen de origen ictal, por lo que se denomina actividad de tipo ictal. En la mayoría se localiza en el lóbulo frontal 9.
Tubulinopatías, lisencefalia y doble corteza
Estudios recientes demostraron que la LIS y la disgiria o malformaciones tipo paquigiria se asocian a mutaciones en los mismos genes y vías principales. De acuerdo a su gravedad se dividen en agiria completa o casi completa (grado 1 y 2), agiria-paquigiria mixta (grado 3); paquigiria (grado 4); paquigiria-heterotopía subcortical en banda (grado 5) y heteropía subcortical en banda (HSB) (grado 6) 4. En las TUB existe ausencia de la laminación cortical, heterotopías radiales y neuronas ectópicas en sustancia blanca. En la LIS, la corteza es de 12-20 mm de espesor y está compuesta por una capa marginal normal, una superficial celular que corresponde a la lámina cortical, una zona de células dispersas y una zona profunda celular con neuronas heterotópicas. Las HSB presentan una corteza normal con 6 capas, una zona delgada de sustancia blanca subyacente a la corteza y una zona densa de neuronas heterotópicas 10. Los genes LIS1 y DCX son los más frecuentemente relacionados a lisencefalia. Las MDC relacionadas con el LIS1 son más graves en áreas posteriores (gradiente posterior-anterior) mientras que las relacionadas al DCX son más graves en áreas anteriores (gradiente anterior-posterior) 4. Alrededor del 60% de los pacientes con LIS y gradiente posterior-anterior son portadores de la mutación en el LIS1 11. Las LIS ligadas al X con agenesia del cuerpo calloso y genitales ambiguos (XLAG) son secundarias a mutaciones en el gen ARX 12. Otros genes relacionados son el RELN o VLDLR, ACTB y ACTG1 6.
Las mutaciones en los genes tubulina fueron primero informadas como causa de lisencefalia (TUBA1A) o PMG (TUBB2B); sin embargo, las TUB comprenden un amplio espectro de anormalidades morfológicas que va desde formas muy extremas de LIS con ausencia completa de giros, agenesia completa del cuerpo calloso e hipoplasia cerebelosa grave, a formas de LIS con moderada a grave hipoplasia cerebelosa; a formas clásicas de LIS y a malformaciones corticales tipo PMG con hipoplasia cerebelosa 4, 8. Se han identificado múltiples genes relacionados a este grupo 13.
Las características neurorradiológicas varían desde una corteza cerebral lisa con ausencia completa de giros a microsurcos tipo PMG. Puede asociarse agenesia del cuerpo calloso y/o hipoplasia cerebelosa. Los pacientes con TUB presentan severos trastornos del neurodesarrollo y epilepsia refractaria. Los espasmos epilépticos son muy frecuentes 14.
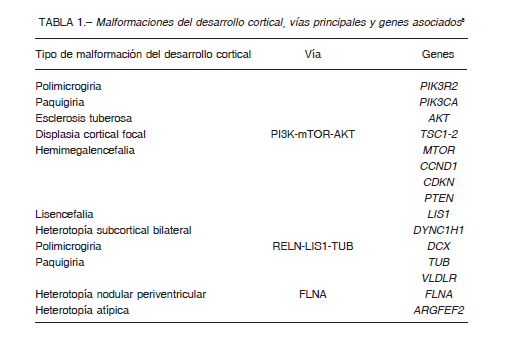
Las manifestaciones clínicas en LIS clásica incluyen retraso mental grave, hipotonía axial, espasticidad, apneas y dificultad en la succión y epilepsia. Las crisis epilépticas comienzan en los primeros tres meses, generalmente se trata de espasmos epilépticos. En niños mayores se observan crisis generalizadas tónicas, atónicas, mioclónicas y espasmos epilépticos periódicos. El EEG muestra ritmos de tipo alfa de elevada amplitud con predominio en las áreas anteriores y se activan durante el sueño como un patrón distintivo e hipsarritmia (Fig. 1A, B, C y D). En pacientes mayores, se observan ritmos rápidos difusos, paroxismos de punta y polipuntas generalizados, descargas de punta-onda bilaterales, variedad lenta y espigas multifocales 9.
Las principales manifestaciones de la HSB son retraso mental y epilepsia (presente en casi todos los pacientes y evolución a la refractariedad en 65%). El 50% tiene convulsiones focales y el restante generalizadas (síndrome de Lennox-Gastaut) 15.
Polimicrogiria
La PMG se caracteriza por pequeñas y excesivas circunvoluciones corticales. Su distribución varía desde
formas difusas a bilaterales, simétricas a bilaterales, asimétricas a unilateral. En ocasiones, puede asociarse con esquizencefalia. La corteza perisilviana es la más frecuentemente afectada 16.
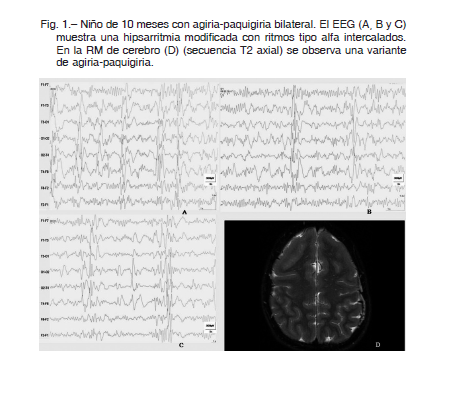
La etiología es variada, existen formas genéticas y adquiridas y también varios tipos de herencia. Puede ser secundaria a citomegalovirus o formar parte de síndromes genéticos como el de DiGeorge, de Stickler y de PHACE (Fig. 2 A, B, C, D y E) 17. Las deleciones del 22q11.2 y 1p36.3 están descriptas. Mutaciones en el PIK3R2 se relacionan con la PMG perisilviana bilateral y formas aisladas de PMG. Se ha descripto recientemente una mutación en el FIG4 en una familia con PMG temporo-occipital y epilepsia 18. La PMG con microcefalia congénita grave se han asociado a mutaciones en el WDR62, o KATNB1 o NDE1 4. La PMG asociada con microcefalia o perímetro cefálico normal ha sido informada en el TUBB2B y el DYNC1H1 8.
Las características neurorradiológicas son corteza de apariencia engrosada (6-8 mm), excepto en los lactantes cuya corteza puede no parecer engrosada debido a la falta de mielinización. Es más frecuente en la región perisilviana uni o bilateral. Estudios de pacientes con PMG con RM 7T han mostrado áreas anormales corticales más extensas que las observadas con técnicas de menor resolución 8.
La clínica es variable. Las formas más graves incluyen microcefalia, examen anormal y heterotopías o hipoplasia cerebelosa. En el síndrome perisilviano existe disfunción oromotora, compromiso intelectual y epilepsia 5. La PMG perisilviana unilateral se presenta frecuentemente con hemiparesia y convulsiones focales. Algunos pacientes con PMG hemisférica unilateral desarrollan epilepsia de difícil control con punta onda continua durante el sueño.
Cuando a la PMG se asocia la esquizencefalia a labio cerrado, los pacientes padecen hemiparesia o retraso
motor, mientras que si es a labio abierto existe hidrocefalia, convulsiones y compromiso cognitivo, el cual puede ser grave. Existen espasmos epilépticos, crisis parciales complejas, tónicas, atónicas y menos frecuentemente tónico-clónicas 4.
En la PMG unilateral la epilepsia está presente en el 80% de los casos y debutan entre el año y los 11 años. Inicialmente son crisis focales con o sin generalización secundaria. Entre los 2 y 9 años ocurren cambios en el patrón de crisis y el EEG y son frecuentes las crisis mioclónicas negativas, con inestabilidad de la marcha, mioclónicas positivas y ausencias atípicas. Al inicio, el EEG interictal muestra espigas focales correspondientes con el área de PMG. Cuando el cuadro clínico cambia, el EEG interictal muestra espigas bilaterales asimétricas durante la vigilia y una actividad simétrica y asimétrica de espiga-ondas continuas durante el sueño lento, que constituye un estado epiléptico eléctrico durante el sueño.
También se observan descargas de espiga-onda de alta frecuencia de descarga bilaterales durante el sueño
lento. El aumento de la frecuencia de las alteraciones interictales en vigilia o sueño, la presencia de espiga o espiga-onda bilaterales simétricas o asimétricas, sincrónicas o asincrónicas o paroxismos de punta-onda bilaterales predominantes en las regiones anteriores son aspectos electroencefalográficos sugestivos del inicio de bisincronías bilaterales secundarias 9, 19.
Heterotopías
La heterotopía es la presencia de neuronas normales en una localización incorrecta. La forma más común es la heterotopía periventricular (HPV), aunque existen otros tipos (subcortical focal o multifocal y leptomeníngea).
Microscópicamente, el tejido heterotópico contiene neuronas y células gliales que se forman en grupos de
nódulos circulares e irregulares separados por capas de fibras mielinizadas. La forma más común de HPV difusa está causada por mutaciones en el gen FLNA, forma ligada al cromosoma X 8 y es frecuente en las formas familiares. Otro gen, el ARFGEF2 está asociado con HPV y microcefalia. Ambos genes regulan las uniones de actina, el tráfico vesicular, la adhesión celular y la función de la glía radial 20.
Las características radiológicas en pacientes con el FNLA1 son bastante homogéneas: nódulos continuos
periventriculares y bilaterales que respetan los cuernos temporales, hipoplasia de vermis y megacisterna magna. En los casos esporádicos el patrón es de predominio posterior. En pacientes con HPV clásica, sin otra MDC, son comunes las convulsiones y el neurodesarrollo puede ir desde normal hasta estar moderadamente comprometido. La mayoría de las mujeres con HPV, debido a mutaciones del FLNA, tienen epilepsia y nivel cognitivo normal o limítrofe. Los fenotipos más graves se han visto con formas atípicas 4.
Conclusiones
Las MDC son una causa muy frecuente de epilepsia y trastornos del desarrollo. El espectro de síndromes epilépticos asociados con las MDC es amplio e incluye desde encefalopatías epilépticas de comienzo temprano a epilepsias focales de debut más tardío. Teniendo en cuenta que la evolución de la epilepsia hacia la refractariedad en las MDC es importante, se requiere de un diagnóstico precoz y la planificación de diferentes estrategias terapéuticas para influir en el pronóstico de los pacientes. La clasificación de las MDC en epilepsia ha evolucionado en este último tiempo gracias al conocimiento de vías moleculares
sumado a las técnicas de neuroimagen, enfatizando la significancia de mecanismos comunes entre el desarrollo cerebral anormal y la epilepsia.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Barkovich J, Dobyns W, Guerrini R. Malformations of cortical development and epilepsy. Cold Spring Harb Perspect Med 2015; 5: a022392.
2. Bae BI, Tietjen I, Atabay KD, et al. Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science 2014; 343: 764-8.
3. André VM, Wu N, Yamazaki I, et al. Cytomegalic interneurons: a new abnormal cell type in severe pediatric cortical dysplasia. J Neuropathol Exp Neurol 2007; 66:491-504.
4. Parrini E, Conti V, Dobyns WB, Guerrini R. Genetic basis of brain malformations. Mol Syndromol 2016; 7: 220-33.
5. Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain 2012; 135: 1348-69.
6. Guerrini R, Dobyns WB. Malformations of cortical development: clinicalfeatures and genetic causes. Lancet Neurol 2014; 13: 710-26.
7. Mirzaa GM, Conway RL, Gripp KW, et al. Megalencephalycapillary malformation (MCAP) and megalencephalypolydactylypolymicrogyria-hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth andabnormal brain and body morphogenesis. Am J Med Genet 2012; 158A: 269-91.
8. Kuzniecky R. Epilepsy and malformations of cortical development: new developments. Curr Opin Neurol 2015, 28: 151-7.
9. Caraballo R, Cersósimo R. Patrones electroencefalográficos particulares en enfermedades y síndromes específicos. En: Caraballo R, CersósimoR, eds. Atlas de Electroencefalografía en la Epilepsia, 1a ed. Buenos Aires: Editorial Panamericana, 2010, p 260-312.
10. Pilz D, Stoodley N, Golden JA. Neuronal migration, cerebral cortical development, and cerebral cortical anomalies. J Neuropathol Exp Neurol 2002; 61: 1-11.
11. Mei D, Lewis R, Parrini E, et al. High frequency of genomic deletion and duplication in the LIS1 gene in lissencephaly: implications for molecular diagnosis. J Med Genet 2008; 45: 355-61.
12. Kato M, Das S, Petras K, et al. Mutations of ARX are associated with striking pleiotropy and consistent genotypephenotype correlation. Hum Mutat 2004; 23: 147-59.
13. Romaniello R, Arrigoni F, Fry AE, et al. Tubulin genes and malformations of cortical development. Eur J Med Genet 2018; 61: 744-54.
14. Romaniello R, Arrigoni F, Bassi MT, Borgatti R. Mutations in α- and β-tubulin encoding genes: implications in brain malformations. Brain Dev 2015; 37: 273-80.
15. Guerrini R, Filippi T. Neuronal migration disorders, genetics, and epileptogenesis. J Child Neurol 2005; 20: 287-99.
16. Kuzniecky R, Andermann F, Guerrini R. Congenital bilateral perisylvian syndrome: study of 31 patients. The CBPS Multicenter Collaborative Study. Lancet 1993; 341: 608-12.
17. Hess CP, Fullerton HJ, Metry DW et al. Cervical and Intracranial Arterial Anomalies in 70 patients with PHACE syndrome. AJNR Am J Neuroradiol 2010; 31: 1980-6.
18. Baulac S, Lenk GM, Dufresnois B, et al. Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria. Neurology 2014; 82: 1068-75.
19. Caraballo RH, Cersósimo RO, Fortini PS, et al. Congenital hemiparesis,unilateral polymicrogyria and epilepsy with or without status epilepticus during sleep: a study of 66 patients with long-term follow-up. Epileptic Disord 2013; 15: 417-27.
20. Sheen VL, Topçu M, Berkovic S, et al. Autosomal recessive form of periventricular heterotopia. Neurology 2003; 60: 1108-12.