
JUAN PABLO APPENDINO 1, JUAN IGNACIO APPENDINO 2
1 Clinical Neuroscience, Department of Pediatrics, Alberta Children’s Hospital, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada, 2 Servicio de Neurología Pediátrica, Departamento de Pediatría, Hospital Italiano de Buenos Aires, Argentina
Resumen Las encefalopatías epilépticas es un grupo de síndromes epilépticos caracterizados por el deterioro cognitivo más allá de lo esperado debido a la actividad epiléptica. Se caracterizan por presentar resistencia farmacológica grave, electroencefalogramas profundamente anormales, inicio en la niñez temprana, deterioro neurocognitivo, fenotipo variable y resonancia magnética de cerebro usualmente normal. Frecuentemente estos síndromes están genéticamente determinados. Su diagnóstico correcto y oportuno puede contribuir y guiar el consejo médico y terapia adecuada, influyendo así en el pronóstico a corto, mediano y largo plazo. En este artículo se revisan los hallazgos electroencefalográficos, genéticos y opciones terapéuticas más recomendadas, facilitando así la conducta clínica. Las encefalopatías epilépticas incluidas en este artículos abarcan los síndromes de Ohtahara, encefalopatia mioclónica temprana, epilepsia focal migratoria de la infancia, West, Dravet, estado mioclónico en encefalopatías no progresivas, Doose, Lennox-Gastaut, Landau-Kleffner y epilepsia con espiga-onda continuas durante el sueño de onda lenta.
Palabras clave: encefalopatía epiléptica, etiología, gen, tratamiento, electroencefalograma
Abstract Genetically determined epileptic encephalopathies. Epileptic encephalopathies is a group of epileptic syndromes characterized by progressive cognitive impairment beyond the expected for the epilepsy activity. They are characterized by severe pharmaco-resistant epilepsy, severely abnormal electroencephalograms, early-age onset, neurocognitve impairment, variable phenotype and usually normal brain MRI. These syndromes are usually genetically determined. A correct and timely diagnosis could help and guide the medical counselling and the correct therapeutic approach improving the short, medium and long term outcomes. In this article we review the electroencephalographic and genetic findings along with the most recommended therapeutic options facilitating the clinical management. We include the following epileptic encephalopathy syndromes: Ohtahara, early myoclonic encephalopathy, epilepsy of infancy with migrating focal seizures, West, Dravet, non-progressive myoclonic status, Doose, Lennox-Gastaut, Landau-Kleffner and continuous spike-wave during sleep epilepsy.
Key words: epileptic encephalopathy, etiology, gene, treatment, electroencephalogram
e-mail: jp.appendino@ahs.ca
En el año 2001 se relacionaron por primera vez las variantes del gen SCN1A con la causa de la encefalopatía epiléptica (EE) denominada síndrome de Dravet 1. La clínica de la EE con inicio neonatal e infantil ha sido reconocida por años, pero el concepto fue formalmente introducido por la Liga Internacional contra la Epilepsia (ILAE, del inglés) recién en el 2001 2 y actualizado en 2010 3. Las EE son epilepsias en las cuales la propia actividad epiléptica podría contribuir al deterioro cognitivo progresivo más allá de lo esperado de la enfermedad original 3.
Gracias a las técnicas moleculares modernas se aceleró el descubrimiento de nuevos genes asociados a EE. Poder reconocer dichas EE es el primer paso para el tratamiento de estas enfermedades. Este articulo trata de resumir los puntos principales de cada EE para facilitar su reconocimiento clínico y neurofisiológico, guiar la pesquisa genética y orientar en la terapéutica
Encefalopatías epilépticas
Ciertos síndromes epilépticos (SE) se asocian comúnmente con un estado encefalopático aunque cualquier síndrome epiléptico podría evolucionar a una EE, si las convulsiones y la actividad interictal son lo suficientemente graves 3. En el año 2017 la ILAE modernizó el concepto de EE e introdujo el de encefalopatía epiléptica del desarrollo (EED) para referirse a pacientes en los cuales la etiología y la gravedad de las convulsiones o actividad eléctrica anormal, son responsables del deterioro cognitivo 4. Se recomiendan los términos “encefalopatía del desarrollo” (ED), “encefalopatía epiléptica” (EE) o EED, según lo que mejor describa al paciente 4.
Tanto un SE reconocido con EE o una EE no específica infantil temprana (EIEE, del inglés) comparten características similares 3-5:
– Epilepsia con resistencia farmacológica grave
– Electroencefalogramas (EEG) difusos y a menudo profundamente anormales
– Presentación en la niñez temprana
– Disfuncionalidad del desarrollo en todos los dominios asociadas a la actividad epiléptica
– Retraso, detención o regresión en el desarrollo.
– Fenotipo variable
– Resonancia magnética (RM) de cerebro frecuentemente normal
Cada EIEE tiene particularidades y generalidades, por ejemplo EIEE2 o desorden con deficiencia de CDKL5 (mutación del gen CDKL5) puede presentar una crisis patognomónica, secuencia “espasmos-tónico-hipermotor”, pero también crisis reflejas 6, 7. Los diferentes fenotipos pueden explicarse debido a mutaciones variadas en varios lugares dentro del mismo gen. También, un mismo SE puede responder a variaciones en distintos genes 3-5.
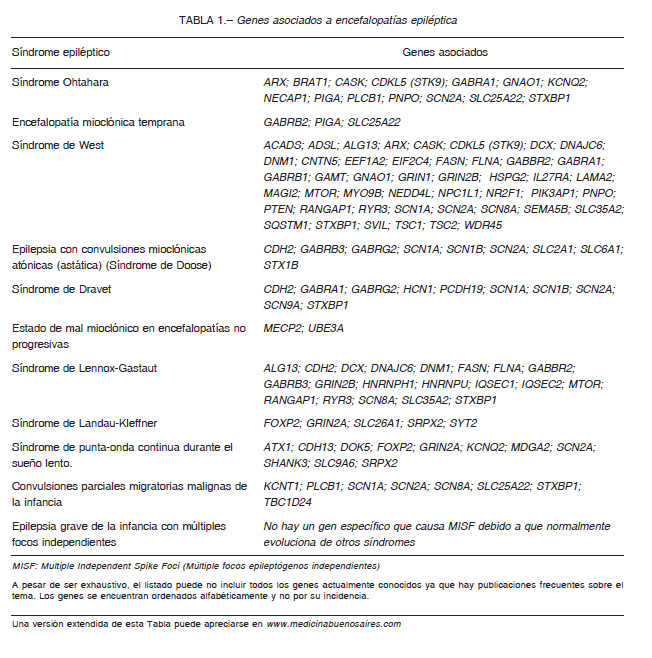
La ILAE incluyó diez SE-EE con etiología genética reconocida, según la edad de comienzo 2, 3, 5, que se desarrollan a continuación. La Tabla 1 resume una lista no exhaustiva de los genes etiológicos y la Figura 1 muestra patrones típicos del EEG.
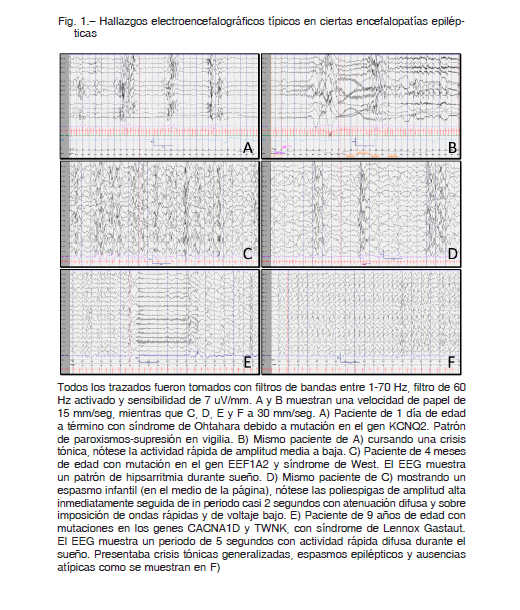
Síndromes epilépticos
Síndrome de Ohtahara 8, 9
Manifestaciones clínicas: Comienzan antes de los 3 meses de edad (frecuentemente antes de los 2 meses).
Se manifiesta con espasmos tónicos cortos pero frecuentes, en vigilia y sueño. Otras crisis como motor focal, generalizada tónico-clónico, se manifiesta en un 30% de los casos. Puede evolucionar a síndrome de West y/o Lennox-Gastaut.
Hallazgos electroencefalográficos: Un patrón de paroxismo-supresión (PS) constante durante el sueño y
vigilia y durante el espasmo-tónico se produce atenuación de la amplitud con actividad rápida.
Enfoque terapéutico: No hay una terapia particularmente efectiva. Se considera la cirugía en hemimegaencefalia o malformaciones cerebrales.
Encefalopatía mioclónica temprana 8, 9
Manifestaciones clínicas: El comienzo es generalmente antes de los 3 meses con mioclonías asincrónicas,
fragmentarias y aleatorias, subcorticales y corticales. El 80% presenta también crisis focales o disautonómicas. Infrecuentemente presentan espasmos tónicos (únicos o en salvas).
Hallazgos electroencefalográficos: El PS es más fácilmente detectable durante el sueño y puede presentarse posteriormente al inicio clínico de mioclonias, evolucionando a veces a hipsarritmia volviendo luego a PS. El PS puede continuar durante toda la infancia.
Enfoque terapéutico: Se debe iniciar piridoxina, o alternativamente, piridoxal-5-fosfato. En casos de hiperglicinemia no cetósica, la indicación es ketamina, triptófano y dextrometorfan más benzoato.
Epilepsia focal migratoria de la infancia 9, 10
Manifestaciones clínicas: Su presentación es generalmente antes de los 6 meses de edad con crisis focales migratorias bilaterales sensoriales o motoras tónico-clónicas. Las crisis sensoriales son difíciles de diagnosticar. Los pacientes pueden desarrollar microcefalia adquirida.
Hallazgos electroencefalográficos: El trazado es normal al comienzo del cuadro, luego ondas lentas focales y posteriormente actividad interictal (punta-onda) multifocal. La actividad ictal es multifocal. Las crisis presentan efecto “ping-pong”, comenzando en un cuadrante cefálico y posteriormente migrando hacia el área homotópica contralateral.
Enfoque terapéutico: Es usualmente refractaria. Algunos casos han respondido a bromuro de potasio pero
luego de más de 3 semanas). Se puede considerar: bromuro de potasio, estiripentol, y levetiracetam; rufinamide y acetazolamida; o estiripentol y clonazepam.
Síndrome de West 9, 11-13
Manifestaciones clínicas: Los espasmos infantiles (EI) se inician entre los 4-24 meses de edad (pico a los 6 meses, y ciertos EIEE presentan un inicio anterior). Los espasmos presentan flexión o extensión axial abrupta tónica de los cuatro miembros, normalmente simétrica y en salvas. Su intensidad es progresiva con disminución gradual (crescendo-decrescendo, o romboidal) y una duración de minutos a más de una hora y frecuentemente al despertar. Los espasmos pueden evolucionar a otro tipo de crisis, en particular motoras tónicas (ver síndrome de Lennox-Gastaut). En los espasmos asimétricos, se debe sospechar lesión cerebral. Los espasmos clínicos pueden aparecer antes que la hipsarritmia en el EEG y al inicio del cuadro hay clara interrupción o regresión del desarrollo, y sociabilización (sonrisa). Solamente el 15-30% de los tratados se normalizan o presentan un ligero retardo (mejor en aquellos sin etiología identificada). El pronóstico mejora cuanto antes se normalice el EEG, y según etiología y tratamiento apropiado. Son factores de pronóstico desfavorable:
– Comienzo < 3 meses
– Presencia varios tipos de crisis
– Persistencia EEG anormal
– RM cerebral anormal
– Duración terapéutica prolongada Hallazgos electroencefalográficos: Hipsarritmia (el 60% presentes durante la vigilia, 100% en sueño).Los espasmos pueden precederla. La hipsarritmia es un trazado desordenado y caótico de ondas lentas de amplitud > 200μV, puntas y punta-onda multifocales, sincrónicas o asincrónicas, ausencia de ritmo posterior dominante y caracteres de sueño. Graduación BASED 12 grado 4 o 5.
Los espasmos: presentan un comienzo de alto voltaje y baja frecuencia (1-2 Hz) con duración de 500 milisegundos, seguido de atenuación difusa con baja amplitud y alta frecuencia (20-30Hz) y duración de 1-5 segundos.
Enfoque terapéutico: Los propósitos terapéuticos son rápida desaparición de los espasmos y eliminación de la hipsarritmia. Hay tres terapias “de primera línea”:
– ACTH (natural o sintética)
– Esteroides orales
– Vigabatrina
Se prefiere utilizar prednisolona (debido a conversión deficiente de prednisona a prednisolona en los niños). Los esteroides orales podrían ser considerados de primera línea debido al costo excesivo de la ACTH y las dudas acerca de su eficacia, excepto en esclerosis tuberosa (se recomienda vigabatrina). También se sugiere utilizar una combinación de esteroides y vigabatrina. Se aplican otras terapias de acuerdo a la etiología: cirugía, dieta cetogénica, vitamina B6, etc.
Síndrome de Dravet 9, 14-16
Manifestaciones clínicas: Su inicio es antes del año de edad en niños previamente normales y evolución
progresiva. La primera convulsión es usualmente prolongada (> 20 min) con fiebre, vacunación en el primer año de vida o baños calientes. Posteriormente presenta convulsiones febriles típicas y atípicas. Luego crisis mioclónicas focales o generalizadas, hemiclónicas o tónico-clónica generalizadas gatilladas por hipertermia o ciertos estímulos, particularmente táctiles, auditivos o visuales. Hay enlentecimiento del desarrollo desde el año hasta los 5 años de edad cuando regularmente se estabiliza. También pueden presentar hipotonía, ataxia, signos piramidales, incoordinación, mioclonías, escoliosis-cifosis y pie plano. El pronóstico es desfavorable y el control total de las crisis es imposible. Cerca de un 20% fallecen por muerte súbita e inesperada en epilepsia (SUDEP, del inglés).
Hallazgos electroencefalográficos: El EEG es normal al comienzo y luego con espiga, poliespigas o punta- onda generalizada o con predominio parieto-central de 4-6 Hz.
Enfoque terapéutico: Se aconseja minimizar baños calientes, ejercicio extenuante y estímulos provocadores en aquellos con convulsiones reflejas. Los bloqueantes de canales de sodio pueden agravar las crisis. Las drogas ácido valproico, clobazam, topiramato, levetiracetam y estiripentol son las de elección. Tanto la Fenfluramina (0.25-1.0 mg/kg/día; max. 20 mg/día) o el cannabidiol (10-20mg/kg/dia) podrían ser beneficiosos.
Estado mioclónico en encefalopatías no progresivas 3, 9, 15
También conocida como “encefalopatías mioclónicas en desórdenes no progresivos” (MSNE, del inglés). Etiológicamente se subdividen en:
– Etiología genética
– Malformación cerebral
– Hipoxia-isquemia.
En este artículo solo abarcaremos el primer grupo.
Manifestaciones clínicas: Abarcan el 48-62% de MSNE. Incluye síndromes de Angelman, Prader-Willi,
Wolf-Hirschhorn, y Rett. Presenta mioclonías muy frecuentes o cuasi-continuas (rítmicas o arrítmicas), ausencias clínicas con mioclonías palpebrales, periorales y distales. También pueden presentar ausencias atípicas, mioclonías masivas, crisis motoras focales, tónicas o clónicas generalizadas. Hay mejoría notable de la conducta al abolir las mioclonías y mejorar el EEG.
Hallazgos electroencefalográficos: Las ausencias mioclónicas con trazado de actividad basal lenta (2-6 Hz) y espiga o poliespiga-onda generalizada de amplitud alta con prevalencia frontal y mioclonía distal errática sobre todo en miembros superiores son frecuentes, seguidas por uno o ambos de los siguientes patrones:
– Breve período silente con actividad lenta (1-5Hz) cuasi-continua a predominio central;
– Breves secuencias de ondas delta rítmicas a predominio parieto-occipital con puntas sobreimpuestas,
frecuentemente activadas por cierre ocular.
Enfoque terapéutico: La combinación de ácido valproico y etosuximida es la más efectiva. La ataxia en el síndrome de Angelman puede empeorar con ácido valproico.
Epilepsia con convulsiones mioclónicas atónicas o síndrome de Doose 9, 17
Manifestaciones clínicas: Se inicia entre los 6 meses y 6 años (pico 3 años), varones 2:1 en niños previamente normales. Frecuentemente es precedido por convulsiones febriles. La primera crisis es usualmente clónica o tónicoclónica generalizada y luego desarrolla crisis mioclónicas-atónicas con vocalización inicial o gruñido (espasmo diafragmático); más tarde caída atónica cefálica o corporal.
La secuencia completa no siempre está presente y pueden presentar ausencias. Remiten en un 80-90% a los 3 años del comienzo y algunos casos experimentan deterioro cognitivo permanente.
Hallazgos electroencefalográficos: Es normal al inicio del cuadro y luego desarrollan breve salvas de 2-5Hz espigas o poliespigas-ondas complejas generalizadas, primariamente durante el sueño y pueden permanecer luego de la mejoría clínica. También actividad occipital puntiaguda de 4Hz atenuada con apertura ocular. EEG ictal muestra espigas o poliespigas-ondas complejas generalizadas seguidas por una atenuación difusa durante la fase atónica.
Enfoque terapéutico: El ácido valproico es de primera elección o la Etosuximida si las ausencias son prominentes.
La dieta cetogénica es de segunda línea. La Lamotrigina podría empeorar las convulsiones mioclónicas.
Síndrome de Lennox-Gastaut 9, 14, 18, 19
Manifestaciones clínicas: Comienza entre los 3-5 años de edad, rango 1-8 años (raramente mayores) y puede
derivar del síndrome de Ohtahara o West. Los trastornos cognitivos o de comportamiento están siempre presentes y pueden preceder a la epilepsia, o ser agravados por la medicación. La triada diagnóstica es:
– Convulsiones de múltiples tipos (tónica obligatoriamente)
– EEG anormal (actividad rápida periódica; PFA, del inglés)
– Trastorno cognitivo
Convulsiones: frecuentemente tónicas generalizadas (más común en sueño), y ausencias atípicas. Otras incluyen tónico-clónicas, atónicas, mioclónicas o mioclónica-atónica.
Es común el estado no convulsivo prolongado.
Hallazgos electroencefalográficos: El PFA es mandatorio para efectuar el diagnóstico, con o sin contracción tónica muscular. Espigas y punta-onda lenta (2-2.5Hz) están presentes en ausencias atípicas. Crisis tónicas pueden presentar espiga-onda generalizada, poliespigas – onda generalizada, atenuación generalizada de voltaje, o actividad de bajo o alto voltaje rápido. El EEG puede evolucionar con el tiempo, mostrando ondas difusas y focales lentas con descargas interictales focales.
Enfoque terapéutico: El ácido valproico es la primera elección. La segunda línea incluye lamotrigina, rufinamida, clobazam, felbamato, topiramato, o zonisamida en tanto que la dieta cetogénica es de tercera línea. Se considera la cirugía cuando es debida en malformaciones cerebrales focales o difusas (VNS, callosotomia, y/o resectiva). Carbamazepina, oxcarbazepina, eslicarbazepina y tiagabina pueden empeorar las crisis. Cannabidiol (10-20mg/kg/dia) y fenfluramina (0.2-0.8 mg/Kg/día) podrían ser beneficiosos.
Síndrome de Landau-Kleffner 9, 20, 21
Manifestaciones clínicas: hay regresión receptiva primero y luego expresiva del lenguaje en niños previamente normales y que pueden comenzar antes de los 2 años de edad con pico entre los 3-6 años. Son infrecuentes las convulsiones generalizadas o focales pero fáciles de controlar. Usualmente la regresión y las convulsiones son concomitantes, pero puede comenzar uno de ellos primero (regresión o convulsiones solamente) seguido del otro. El 30% de los pacientes presenta hiperactividad y agresividad.
Hallazgos electroencefalográficos: Es usualmente normal en vigilia pero en el sueño presenta espigas centrotemporales o más amplias, con distribución bilateral o con predominio del hemisferio dominante. Son continuas durante el sueño de onda lenta (estado epiléptico eléctrico del sueño [ESES, del inglés]).
Enfoque terapéutico: El objetivo es controlar las convulsiones y mejorar el EEG de forma rápida. Los
bloqueadores de canales de sodio podrían agravar el EEG. La transección múltiple subpial no es aconsejada, ya que su pronóstico es similar al tratamiento médico. Los corticoides orales o benzodiacepinas en altas dosis son la primera línea. Las combinaciones de ácido valproico, levetiracetam y etosuximida pueden ser efectivas en tanto que la Amantadina podría ser útil.
Epilepsia con espiga-onda continuas durante el sueño de onda lenta 9, 20, 21
Manifestaciones clínicas: Comienzo es entre los 2 meses y 14 años de edad (pico 4-5 años). Presentan trastorno cognitivo y de comportamiento pero no de lenguaje. Las convulsiones son infrecuentes y predominantemente nocturnas, incluyendo estatus epiléptico. Las crisis tónicas son extremadamente inusuales. Puede evolucionar de una epilepsia benigna con espigas centro-temporales (BECTS, del inglés).
Hallazgos electroencefalográficos: El EEG presenta espiga-onda continuas o cuasi-continuas, usualmente
difusa, 1.5-3 Hz en sueño NREM. Requiere más del 85% del EEG en sueño NREM ocupado con espiga-onda. Algunos autores proponen > 50% del EEG en sueño NREM o una clara activación en NREM comparado con vigilia. Las convulsiones disminuyen y el EEG mejora durante la pubertad pero los hallazgos neuropsicológicos permanecen.
Enfoque terapéutico: Similar al síndrome de Landau-Kleffner. Primera línea: corticoides orales. Benzodiacepinas en altas dosis, combinaciones de ácido valproico, levetiracetam y etosuximida y también Amantadina.
Conclusiones
El reconocimiento y el diagnóstico adecuado de las encefalopatías epilépticas son desafiantes. La utilización de herramientas diagnósticas como el EEG o el estudio genético facilitan la caracterización sindrómica adecuada para iniciar el tratamiento oportuno mejorando así el pronóstico. Se encuentran en desarrollo nuevos tratamientos y estrategias diagnósticas que necesitan validación a gran escala.
Agradecimientos: Los autores agradecen los inagotables esfuerzos realizados por Yanina Eberhard en el tipiado y traducción de este artículo. Sin su ayuda, no se habría terminado en tiempo y forma.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodiumchannel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001; 68: 1327-32.
2. Engel J Jr. International League Against Epilepsy (ILAE). A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001; 42: 796-803.
3. Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 2010; 51: 676-85.
4. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017; 58: 512-21.
5. Engel J Jr. Report of the ILAE classification core group. Epilepsia 2006, 47: 1558-68.
6. Klein KM, Yendle SC, Harvey AS, et al. A distinctive seizure type in patients with CDKL5 mutations: Hypermotor-tonicspasms sequence. Neurology 2011; 76: 1436-8.
7. Peikes T, Hartley JN, Mhanni AA, Greenberg CR, Appendino JP. Reflex seizures in a patient with CDKL5 deficiency disorder. Can J Neurol Sci 2019; 46: 482-5.
8. Beal JC, Cherian K, Moshe SL. Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Pediatr Neurol 2012; 47: 317-23.
9. Fernandez-Concepcion O, Lopez-JimenezM. Epileptic encephalopathies in infants and children. IntechOpen, DOI: 10.5772/intechopen.85378. En: https: //www.intechopen.com/online-first/epileptic-encephalopathies-in-infants-andchildren; consultado junio 2019.
10. Coppola G. Malignant migrating partial seizures in infancy: an epilepsy syndrome of unknown etiology. Epilepsia 2009; 50 Suppl 5: 49-51.
11. Yuskaitis CJ, Ruzhnikov MRZ, Howell KB, et al. Infantile spasms of unknown cause: predictors of outcome and genotype-phenotype correlation. Pediatr Neurol 2018; 87: 48-56.
12. Mytinger JR, Hussain SA, Islam MP, et al. Improving the inter-rater agreement of hypsarrhythmia using a simplified EEG grading scale for children with infantile spasms. Epilepsy Res 2015; 116: 93-8.
13. O’Callaghan FJ, Edwards SW, Alber FD, et al. Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open-label trial. Lancet Neurol 2017; 16: 33-42.
14. Samanta D. Cannabidiol: a review of clinical efficacy and safety in epilepsy. Pediatr Neurol 2019; 96: 24-9.
15. Elia M. Myoclonic status in nonprogressive encephalopathies: an update. Epilepsia 2009; 50 Suppl 5: 41-4.
16. Polster T. Individualized treatment approaches: Fenfluramine, a novel antiepileptic medication for the treatment of seizures in Dravet syndrome. Epilepsy Behav 2019; 91: 99-102.
17. Nickels K, Thibert R, Rau S, et al. How do we diagnose and treat epilepsy with myoclonic-atonic seizures (Doose syndrome)? Results of the Pediatric Epilepsy Research Consortium survey. Epilepsy Res 2018; 144: 14-9.
18. Cross JH, Auvin S, Falip M, Striano P, Arzimanoglou A. Expert opinion on the management of Lennox-Gastaut syndrome: treatment algorithms and practical considerations. Front Neurol 2017; 8: 505.
19. Lagae L, Schoonjans AS, Gammaitoni AR, Galer BS, Ceulemans B. A pilot, open-label study of the effectiveness and tolerability of low-dose ZX008 (fenfluramine HCl) in Lennox-Gastaut syndrome. Epilepsia 2018; 59: 1881-8.
20. van den Munckhof B, Alderweireld C, Davelaar S, et al. Treatment of electrical status epilepticus in sleep: Clinical and EEG characteristics and response to 147 treatments in 47 patients. Eur J Paediatr Neurol 2018; 22: 64-71.
21. Wilson RB, Eliyan Y, Sankar R, Hussain SA. Amantadine: A new treatment for refractory electrical status epilepticus in sleep. Epilepsy Behav 2018; 84: 74-8.