
MARTA MARTÍNEZ-MORGA 1, MARI PAZ QUESADA 1, CARLOS BUENO 2, SALVADOR MARTÍNEZ 2
1Departemento de Anatomía y Psicobiología. IMIB-Arrixaca. Univ. de Murcia. Nurcia. España
2Intituto de Neurociencias UMH-CSIC. San Joan d’Alacant. Alicante. España
Resumen Los trastornos del espectro autista (TEA) son una alteración funcional de la corteza cerebral, que presenta anomalías estructurales del neurodesarrollo que afectan fundamentalmente a la función sináptica y el patrón de conexiones dentro y entre columnas corticales. Desde su aspecto etiológico, el TEA tiene una importante carga genética, considerándose un desorden derivado de una combinación de mutaciones “de novo”, asociadas a una predisposición derivada de variaciones comunes heredadas. Las principales anomalías genéticas asociadas a TEA implican genes que codifican proteínas de la sinapsis. Así, en pacientes con TEA se han descrito alteraciones del desarrollo inicial de las sinapsis en los circuitos de conexión entre áreas corticales de procesamiento complejo. La complejidad molecular observada en la predisposición a desarrollar un TEA, junto con la diversidad de fenotipos estructurales neuronales, ha hecho que los modelos animales reproduzcan solo parcialmente el TEA. Para avanzar en el estudio experimental se hace pues necesario desarrollar modelos más representativos, como son los modelos celulares derivados de células humanas. En las últimas décadas, el desa- rrollo de la biología de las células madre nos da medios para acceder a paradigmas experimentales sobre células derivadas de individuos con TEA. Actualmente, los modelos de células plutipotentes inducidas (IPs) derivadas de células humanas permiten profundizar en el estudio de las bases moleculares y celulares del TEA. Sin embargo, presentan problemas inherentes derivados de la manipulación experimental que conlleva la reprogramación de la expresión génica, por lo que otros modelos celulares se están también postulando como válidos.
Palabras clave: autismo, TEA, neurodesarrollo, sinaptogénesis, modelos celulares de TEA
Abstract Neurobiological bases of autism and cellular models for its experimental study. Autism Spectrum Disorders (ASD) are a functional alteration of the cerebral cortex, which presents structural neuro- developmental anomalies that affect synaptic function and the pattern of connections within and between corti- cal columns. From its etiological aspect, ASD has an important genetic load, considering a polygenic disorder, derived from a combination of “de novo” genetic mutations, associated to a predisposition derived from common inherited variations. The main genetic anomalies associated with ASD involve genes that encode proteins of the synapse. Thus, in patients with ASD, alterations in the initial development of the synapses have been described in the connection circuits between complex processing cortical areas. The molecular complexity observed in the predisposition to develop an ASD, together with the diversity of structural phenotypes, has made animal models reproduce only partially the ASD. To advance in the experimental study it is therefore necessary to develop rep- resentative models, such as cellular models derived from human cells. In recent decades, the advances in stem cell biology give us a way to apply experimental paradigms in cells derived from individuals with ASD. Currently, induced pluripotent cells (IPs) derived from human adult cells allow deepening the study of molecular and cellular bases of the neuronal development in humans, as well as the anomalies in this development, which give rise to disorders such as ASD. However, they present inherent problems derived from the experimental manipulation that involves the reprogramming of gene expression, therefore other models are also been explored.
Key words: autism, ASD, neurodevelopment, synaptogenesis, cellular models of ASD
Dirección postal: Salvador Martinez. Instituto de Neurociencias UMH- CSIC. Avda. Ramón y Cajal. Campus de San Juan. 03550. Sant Joan d’Alacant. España.
e-mail: smartinez@umh.es
El término autismo lo utilizó por primera vez el psi- quiatra Leo Kanner en 1943 en su artículo Autistic Disturbances of Affective Contact, en el que reflejó sus observaciones en 11 niños que mostraban “incapacidad para relacionarse de forma normal con las personas y maduración del sistema nervioso central, que se inician trayectorias evolutivas y, en su manifestación fenotípica se caracteriza por deficiencias persistentes en la comunicación e interacción social en diversos contextos, unidas a patrones restrictivos y repetitivos de comportamiento, intereses o actividades2. En los últimos cuarenta años la prevalencia de TEA se ha incrementado significativamente, situándose en la actualidad en 1 caso por cada 100 nacimientos3.
Bases biológicas del TEA
Las funciones mentales son el resultado de la actividad de las neuronas que constituyen el sustrato celular más especializado del cerebro. Por lo tanto, entender el desarrollo de la estructura y función neuronal es necesario para poder explicar las anomalías que producen trastornos del neurodesarrollo, como es el caso del TEA, y abordar un tratamiento adecuado de sus consecuencias. La riqueza estructural y funcional del cerebro depende del desarrollo de regiones cerebrales especializadas funcionalmente con tipos de neuronas característicos, que establecen precisos patrones de conexiones entre ellas. Esto requiere la articulación en el espacio y el tiempo de los procesos moleculares y celulares que construyen la estructura del sistema nervioso central.
Gracias al progreso de la biología molecular y la secuenciación de genomas completos se están empezando a conocer los procesos por los que la información genética regula el desarrollo cerebral. Aproximadamente, la mitad de las instrucciones de nuestro genoma se dedica a la construcción del cerebro. Se sabe que mediante mecanismos de activación y represión mutua el patrón espacio-temporal de expresión, es decir, cuándo y dónde se expresan los genes, genera interacciones que codifican la forma del embrión y de su cerebro. El desarrollo normal depende, pues, de la información que está escrita
en los genes y del equilibrio de su expresión en cantidad, tiempo y espacio. Los trastornos del neurodesarrollo están asociados a anomalías estructurales derivadas de alteraciones durante el desarrollo embrionario del cerebro, y su consecuencia son desviaciones funcionales que se manifiestan tempranamente en la vida, con la aparición de alteraciones en la conducta, discapacidad intelectual y retraso en el desarrollo psicomotor.
Bases estructurales
El desarrollo neurológico culmina con la madurez funcional del cerebro. Discurre desde la vida fetal hasta el inicio de la edad adulta, desde los estadios embrionarios hasta la poda sináptica que ocurre en la adolescencia y la mielinización, que finaliza al final de la pubertad. Al nacer, el cerebro inmaduro es influenciado por los estímulos ambientales que pueden modificar la expresión de los genes. Esta interacción gen-ambiente (GxA) puede ser plasticidad sináptica se consigue modulando la expresión de un conjunto de genes que regulan mecanismos moleculares y celulares que regulan la dinámica de las conexiones sinápticas. La neuroplasticidad durante el desarrollo del cerebro presenta patrones temporales heterogéneos, existiendo un periodo crítico de mayor maleabilidad sináptica alrededor del nacimiento, que modula la regulación génica para la formación y consolidación de conexiones neuronales adecuadas mediante la influencia de los estímulos ambientales.
Los estudios de neuroimagen del cerebro humano han sido muy útiles para correlacionar fenotipos de conducta con alteraciones en estructuras cerebrales. En el TEA, los datos de resonancia magnética estructural y funcional detectan
la presencia de anomalías estructurales en varios circuitos neuronales en regiones del cerebro social, entre los que se incluyen: la amígdala, los ganglios basales (núcleo acumbens) y corteza prefrontal. Se piensa que son las alteraciones en la corteza prefrontal, y en especial su conexión con la amígdala cerebral y la corteza parietal y temporal, las que se presentan de manera más constante en los estudios realizados en muestras cerebrales humanas y en modelos animales de TEA6.
Diferentes modelos han sido propuestos de alteraciones en la función de los circuitos neuronales, desde una sobreconectividad derivada de una disminución en la poda sináptica hasta una excesiva inhibición colateral retroactiva
entre circuitos corticales9. Casanova y colaboradores han demostrado la presencia de alteraciones estructurales
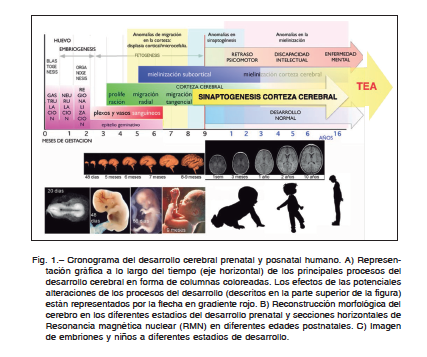
en la corteza cerebral de pacientes con TEA, describiendo un incremento de micro-columnas corticales, con neuro- nas más pequeñas, hiper-excitabilidad intracolumnar y disminución de las conexiones largas de las neuronas corticales10. Estas alteraciones están presentes sobretodo en la corteza prefrontal, posiblemente debido a un desarrollo tardío de esta región, que se extiende durante los primeros años de la infancia. Estas anomalías en la distribución de las neuronas corticales son consecuencia de alteraciones en la proliferación y migración celular durante el desarrollo cerebral, que pueden ser debidas a modificaciones genéticas o exposición a tóxicos que afectan a las células germinales neurales. La displasia cortical y la hiperexcitabilidad microcolumnar explicarían la relación entre TEA y epilepsia11.
En conclusión, desde el aspecto neurobiológico, el TEA es una alteración funcional de la corteza cerebral, que presenta anomalías estructurales en la disposición de las neuronas, así como en la función sináptica y en el patrón de conexiones dentro y entre columnas corticales12. Estas alteraciones afectan fundamentalmente a la corteza prefrontal y sus conexiones, la principal región encefálica implicada en la regulación de la conducta social. A nivel funcional, estas alteraciones producen anomalías en el proceso de neuroplasticidad del desarrollo, como ocurre en otros trastornos neuropediátricos congénitos y adquiridos, como la encefalopatía por hipoxia neonatal, parálisis cerebral, epilepsia, distonía y discapacidad intelectual; así como en la esquizofrenia.
Bases genéticas
Desde su aspecto etiológico, ya hemos visto cómo el TEA tiene una importante carga genética, considerándose un desorden poligénico (múltiples genes implicados con carga patogénica escasa y variable) y, por lo tanto, consecuencia de una combinación de alteraciones genéticas de novo (mutaciones espontáneas) asociadas a una predisposición derivada de variaciones comunes heredadas.
Las principales anomalías genéticas asociadas a TEA implican genes que codifican proteínas de la sinapsis13. Entre ellas, proteínas que regulan la transcripción del ADN o la traducción del ARNm, proteínas del citoesqueleto y de membrana, de conexiones intercelulares y de maduración celular. En general, son genes ligados al establecimiento y mantenimiento de las sinapsis y a la plasticidad sináptica que ocurre en el aprendizaje. Como hemos descrito anteriormente, en pacientes con TEA se han descrito alteraciones del desarrollo inicial de las sinapsis en los circuitos de conexión entre áreas corticales de procesamiento complejo (que reciben y procesan de forma combinada información multimodal), sobre todo de los lóbulos frontal, temporal y parietal14.
Bases moleculares del TEA
En relación con las bases genéticas del TEA, en los últimos años se han postulado, al menos, dos mecanismos patogé- nicos diferentes de predisposición a padecer este trastorno:
– Heterogeneidad genética derivada de la combinación de múltiples variantes comunes, es decir, hasta 150 genes con diferente localización cromosómica.
– Un modo poligénico de herencia, con alteraciones raras de pocos genes, que requiere la combinación de anomalías en varios de estos genes.
Aunque hoy en día se tiende a favorecer el modelo poligénico, ambos modelos indican que las interacciones entre genes (GxG) pueden tener un importante papel patogénico, es decir, la alteración en la expresión de un gen puede depender de variaciones en la secuencia de otro gen15.
En el marco de esta heterogeneidad genética, los estudios moleculares pueden proporcionar la información necesaria para proponer hipótesis explicativas de las re- laciones GxG; así como de las relaciones GxA en el TEA.
Las proteínas relacionadas con el desarrollo neural y la función sináptica son de especial relevancia para conocer las bases moleculares de la patogénesis del TEA16, siendo estas las que hemos priorizado para ser estudiadas en nuestro trabajo.
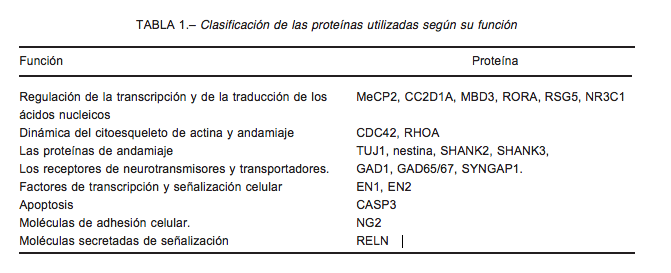
Las proteínas identificadas en la literatura con un papel en la fisipatología del TEA pueden distribuirse en ocho grupos según la función celular en la que interviene (Tabla 1).
Importancia de los factores ambientales
El origen genético del TEA esta abiertamente aceptado, pero existen evidencias que apoyan la contribución de factores ambientales y epigenéticos en su desarrollo. Los factores genéticos y ambientales juegan un papel determi- nante en el riesgo de padecer TEA. La identificación de las etapas del desarrollo en las que existe más vulnerabilidad es de gran importancia para la comprensión de cuándo y en qué circunstancias un niño está en riesgo elevado de desarrollar un TEA. No existe un factor ambiental que, en exclusiva, explique el aumento de la prevalencia del autismo. Sin embargo, este aumento en las últimas décadas podría deberse a factores ambientales y al estilo de vida, que afectarían las interacciones entre el ambiente y la expresión genética en el desarrollo del niño, tanto en el útero materno como en la etapa postnatal. Los factores de riesgo que se han identificado son: agentes infecciosos, medicamentos, sustancias químicas ambientales, dieta y estrés físico/psicológico4-6, 8.
Mecanismos de interacción GxA: epigenética
El término “epigenética” se refiere a los cambios que se producen en la cromatina que, aunque no modifican la secuencia de los nucleótidos del ADN, sí que dirigen la expresión de los genes, manteniendo un equilibrio entre la formación de eucromatina (transcripcionalmente activa, laxa y accesible) y de heterocromatina (trancripcional- mente más inactiva, menos accesible) en respuesta a los requerimientos celulares. Las modificaciones epigenéticas incluyen la metilación del ADN, las modificaciones post- transduccionales de las histonas, los pequeños ARNs no codificantes (como los microARNs) y los complejos proteicos remodeladores de cromatina. Son estables, con frecuencia heredables en la división celular y solo son ocasionalmente heredadas por la descendencia en el proceso de formación de los gametos sexuales o meiosis17. Estos factores epigenéticos están involucrados directamente en el desarrollo y en el envejecimiento del ser humano, mediante su participación en procesos como la diferenciación celular, la impronta genómica, la inactivación del cromosoma X y la expresión de genes específicos de tejido. De esta manera, modificaciones epigenéticas aberrantes pueden alterar la expresión de estos genes diana, y dar lugar a diferentes desordenes del neurodesarrollo17.
La complejidad molecular observada en la predisposición a desarrollar un TEA, sobretodo aquella que regula el establecimiento y la función de los contactos sinápticos, junto con la diversidad de fenotipos estructurales neuro-
nales, ha hecho que los modelos animales reproduzcan
solo parcialmente el TEA. Por lo tanto, para avanzar en el estudio experimental es necesario desarrollar modelos más representativos, como son los modelos celulares derivados de células humanas. En las últimas décadas, el desarrollo de la biología de las células madre nos ha permitido acceder a paradigmas experimentales sobre células derivadas de individuos con TEA.
Modelos celulares de TEA
En el año 2006 fueron generadas las células madre pluripotenciales inducidas (IPs). Las IPs son células adultas diferenciadas, pero tras la inducción con la inserción de 4 genes en su núcleo (Oct4, Sox2, c-Myc, y Klf4), se comportan como células embrionarias con plasticidad histogenética. Estas células han supuesto un nuevo enfoque en la biología y terapia celular18, por permitir disponer de células con características pluripotenciales sin tener que manipular o destruir embriones. Estas células IPs son morfológicamente similares a las células madre embrionarias, expresan genes y antígenos de superficie celular idénticos a éstas y tienen capacidad de formar teratomas cuando se trasplantan en ratones inmunodeprimidos. Estas células son inmortales y pueden reprogramarse para tipos de células diferenciadas, incluidas las células cerebrales.
Las IPs se han utilizado para estudiar los trastornos del neurodesarrollo, ya que estas células permiten generar modelos celulares de trastornos del neurodesarrollo humano in vitro y, de hecho, reconstruir la trayectoria alterada del desarrollo cerebral como se ve en trastornos como el TEA. Los fibroblastos obtenidos a partir de una biopsia cutánea de cualquier individuo se pueden “desprogramar” en células madre pluripotentes inducidas por el ser humano. Estas IPs derivadas de células humanas se pueden diferenciar en progenitores neuronales y neuronas más maduras in vitro, permitiendo así la generación de modelos de trastornos neurológicos específicos del paciente. El laboratorio de la doctora Vaccarino ha de- mostrado que las IPs derivadas de pacientes conservan la firma genética única de los pacientes de los que se derivaron originalmente19. Estas células pueden ayudar a evaluar los efectos de alterar la expresión de genes específicos (conocidos o desconocidos) en el desarrollo típico de las células neuronales humanas.
Hasta ahora, nuestro conocimiento y comprensión de los trastornos del neurodesarrollo se ha limitado en gran medida a los hallazgos de los estudios de asociación del genoma completo y los modelos de roedores. Si bien, como hemos señalado anteriormente, los modelos de roedores proporcionan información sobre los procesos fundamentales y evolutivamente conservados, que subyacen al desarrollo neuronal, no proporcionan información precisa sobre la patogénesis de la enfermedad humana y no son idóneos para testar de forma clara las vías terapéuticas. Esto se debe a la mayor complejidad funcional, tamaño y diversidad de tipos celulares encontrados en el cerebro humano. Actualmente, los modelos de células IPs derivadas de células humanas nos presentan un medio poderoso para profundizar en las bases moleculares y celulares del desarrollo del cerebro humano, así como las anomalías en este desarrollo, que dan lugar a trastornos como el TEA.
Otros modelos potencialmente útiles: las células madre de origen dental
La formación embriológica de los dientes, se caracteriza por una estructurada interacción de células ectodérmicas epiteliales y ectomesénquima originado por la migración de las células de la cresta neural. Este tejido origina la pulpa dental (PD) y el ligamento periodontal (LP) tanto de leche como permanentes. El análisis inmunohistoquímico de estas células mostró marcadores similares a las células mesenquimales extraídas de médula ósea.
En el caso de las células madre adultas en el LP son similares a otras células madre mesenquimales con respecto a la expresión de STRO-1, CD146, CD90, CD29, CD44, CD13, CD105 y CD166, por lo que las células madre del LP pueden también derivar de una población de células perivasculares que conservan capacidad autorregenerativa y multipotencialidad. Se ha investigado el potencial de células aisladas del LP para dar lugar a células propias del sistema nervioso. Para ello células aisladas del LP humano han sido cultivadas con diferentes medios descritos para células madre del sistema nervioso. Cuando el medio estaba libre de suero y con EGF Y FGF2, éstas se han propagado como agregados celulares que recuerdan a las neuroesferas, modo por el cual proliferan las CMN. Este hecho refuerza la importancia de las células de la cresta neural en el desarrollo y capacidad regenerativas de las piezas dentarias. Al analizarlas se descubrió que expresaban marcadores de células madre neurales como Nestina y Sox-2, además de Emx2, un marcador temprano del desarrollo del neocortex, y A2B5, un antí- geno neuronal de superficie. Si son tratadas con ácido retinoico, adquirían una morfología neuronal y llegaban a expresar marcadores neuronales como la β-III-tubulina, neurofilamento-M, neurofilamento-H, MAP2, GAD67, neurofilamento-L y sinaptofisina. Se ha demostrado su potencialidad neural tanto “in vitro” como “in vivo”, así como la expresión de marcadores específicos de progenitores de cresta neural, como Slug, Twist y Sox920.
Los principales componentes del tejido de la pulpa dental son células mesenquimales derivadas de la cresta neural. Algunas de estas células demuestran un alto potencial de crecimiento y poseen múltiples propiedades de diferenciación. Se ha aislado una población de células madre mesenquimales (MSC) a partir de tejido de pulpa dental.
Los modelos celulares derivados de tejidos humanos están abriendo nuevos horizontes para poder desarrollar diseños experimentales más adecuados en el estudio de las bases moleculares y celulares del TEA.
Agradecimientos: Este trabajo ha sido financiado por los siguientes proyectos de investigación: FEDER BFU2011-27326, SAF2014-59347-C2-1-R and Severo Ochoa Excellence Project SEV-2013-0317; Instituto de Salud Carlos III: Red TERCEL RD12/0019/0024, Generalitat Valenciana: PROME- TEO II/2014/014.
Conflicto de intereses: No existe conflicto de intereses para ningún autor del artículo.
Bibliografía
1. twins among affected sibling pairs with autism: implications for the etiology of autism. Amer J Human Genetics 2001; 69: 1062-7.
2. Xavier J, Bursztejn C, Stiskin M, Canitano R, Cohen D. Autism spectrum disorders: An historical synthesis and a multidimensional assessment toward a tailored thera- peutic program. Res Autism Spectr Dis 2015; 18: 21-3.
3. Elsabbagh M, Divan G, Koh YJ, et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res 2012; 5: 160-79.
4. Dierssen M, Martinez S. Neuropathology and synaptic al- terations in neurodevelopmental disorders. En: Caheney DS, Sklar P, Buxbaum JD, Nestler EJ, eds. Neurobiology of mental illness. 4th. ed. Oxford: Oxford University Press; 2013, p 980.
5. Martinez-Morga M, Martinez S. Brain development and plasticity. Rev Neurol 2016; 62 Suppl 1: S3-8.
6. Martinez-Morga M, Quesada-Rico MP, Bueno C, Martinez S. Neurobiological bases of autistic spectrum disorder and attention deficit hyperactivity disorder: neural dif- ferentiation and synaptogenesis. Rev Neurol 2018; 66 (S01): S97-S102.
7. Ismail FY, Fatemi A, Johnston MV. Cerebral plasticity: Windows of opportunity in the developing brain. Eur J Paediatr Neurol 2017; 21: 23-48.
8. Martinez-Morga M, Martinez S. Neuroplasticity: synaptogenesis during normal development and its implication in intellectual disability. Rev Neurol 2017; 64 (s01): S45- S50.
9. Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 2005; 6: 889-98.
10. Casanova MF, El-Baz AS, Kamat SS, et al. Focal cortical dysplasias in autism spectrum disorders. Acta Neuro- pathol Commun 2013;1: 67.
11. Strasser L, Downes M, Kung J, Cross JH, De Haan M. (2017). Prevalence and risk factors for autism spectrum disorder in epilepsy: a systematic review and meta- analysis. Dev Med Child Neuro 2017; 60: 19-29.
12. Batshaw ML, Towbin KE. Research news: the origings of autism. Pediatric Res 2001; 50: 1-2.
13. Sala C, Verpelli C. Neuronal and Synaptic Dysfunction in Autism Spectrum Disorder and Intellectual Disability. 1st Edition. Academic Press; ISBN: 9780128001097, 2016, 394 p.
14. Hutsler JJ, Casanova MF. Review: Cortical construction in autism spectrum disorder: columns, connectivity and the subplate. Neuropathol Appl Neurobiol 2016; 42:15-34.
15. Skafidas E, Testa R, Zantomio D, Chana G, Everall IP, Pantelis C. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Molecular Psy- chiatry 2014; 19, 504-10.
16. Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci 2006; 29: 349-58.
17. LaSalle JM. A genomic point-of-view on environmental factors influencing the human brain methylome. Epigenetic 2011; 6: 862-9.
18. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126: 663-76.
19. Abyzov A, Mariani J, Palejev D, Zhang Y, Haney MS, Tomasini L. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 2012; 492: 438-42.
20. Bueno C, Ramirez C, Rodriguez-Lozano FJ, et al. Human adult periodontal ligament-derived cells integrate and differentiate after implantation into the adult mammalian brain. Cell Transplant 2013; 22: 2017-28.