
ANA I. DOWNEY 1, VERÓNICA CORTÉS GUERRERI 2, ROBERTO D. FREUE 1, ANA V. LUDUEÑA 1
1 Servicio de Clínica Médica, 2 Servicio de Hematología, Instituto de Investigaciones Médicas Alfredo Lanari, Buenos Aires, Argentina
Resumen El síndrome de lisis tumoral (SLT) es una entidad poco frecuente y potencialmente fatal. Representa una emergencia oncológica. Puede diagnosticarse por su forma de presentación clínica y también por los resultados de laboratorio. En la mayoría de los casos se presenta como complicación del tratamiento quimioterapéutico de enfermedades oncohematológicas con gran masa tumoral. Con menor frecuencia se ha descrito un síndrome de lisis tumoral espontáneo, o secundario al uso de corticoides, hidroxiurea y radioterapia. En sus formas más graves puede requerir internación en unidades de terapia intensiva y medidas terapéuticas invasivas como la hemodiálisis. Comunicamos cuatro casos de SLT con características de presentación inusual internados en nuestro Instituto de Investigaciones Médicas.
Palabras clave: síndrome de lisis tumoral, insuficiencia renal aguda, quimioterapia
Abstract Tumor lysis syndrome (SLT) is a rare and potentially fatal entity. It represents an oncological emergency. It can be diagnosed by its clinical presentation and also by laboratory results. In most cases it is presented as a complication of the chemotherapeutic treatment of oncohematological diseases with large tumor mass. Less frequently, a syndrome of spontaneous tumor lysis has been described, or secondary to the use of corticosteroids, hydroxyurea and radiotherapy. In its most severe forms it may require hospitalization in intensive care units and invasive therapeutic measures such as hemodialysis. We report four cases of SLT with unusual presentation characteristics admitted to our Medical Research Institute.
Key words: tumor lysis syndrome, acute renal failure, chemotherapy
Dirección postal: Ana V. Ludueña, Instituto de Investigaciones Médicas Alfredo Lanari, Combatientes de Malvinas 3150, 1427 Buenos Aires, Argentina
e-mail: ana_luduena@hotmail.com
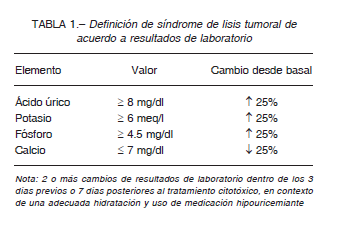
El síndrome de lisis tumoral (SLT) es una entidad infrecuente. Representa una emergencia oncológica potencialmente fatal. Según la clasificación de Cairo y Bishop, puede definirse un SLT de laboratorio (Tabla 1) y otro clínico 1. El diagnóstico de SLT clínico se realiza con la presencia del SLT de laboratorio y la suma de al menos una de las siguientes manifestaciones: aumento de la creatinina, convulsiones, arritmias cardíacas o muerte 1.
La mayor parte de la información sobre esta entidad proviene de publicaciones internacionales retrospectivas y de comunicaciones de casos, que tienden a reflejar una baja incidencia de SLT clínico 1, 2. Es usualmente una complicación del tratamiento de enfermedades oncohematológicas con gran masa tumoral, tumores con alta sensibilidad a la quimioterapia y alta tasa de replicación.
Con menor frecuencia se ha descrito al síndrome de lisis tumoral espontáneo (SLTE), o secundario al uso de corticoides, hidroxiurea y radioterapia 2.
Los factores de riesgo para su desarrollo están relacionados a las características propias de la enfermedad (gran tamaño tumoral, compromiso de otros órganos, infiltración de médula ósea, quimiosensibilidad y tasa de replicación) y al estado del paciente, como la presencia de deshidratación o insuficiencia renal previa 2, 3.
Las manifestaciones clínicas son la consecuencia de alteraciones hidroelectrolíticas que se generan por la destrucción de células malignas ricas en potasio, fósforo y ácido úrico, a las 24-48 h del inicio de la quimioterapia. Las complicaciones sistémicas pueden incluir arritmias cardíacas, convulsiones, tetania e insuficiencia renal aguda.
Su prevención implica la hiperhidratación parenteral (2-3 l/m2 de fluidos por día) con soluciones cristaloides para aumentar la perfusión y la filtración glomerular, y el uso de fármacos que disminuyen los niveles plasmáticos de ácido úrico, como alopurinol (inhibidor de la xantinoxidasa) y rasburicasa (urato-oxidasa recombinante) 1, 2. El tratamiento del SLT establecido involucra, además de la hidratación parenteral y el uso de medicación hipouricemiante, el control específico de las alteraciones hidroelectrolíticas y sus complicaciones, incluyendo eventualmente el reemplazo de la función renal. Algunos pacientes desarrollan insuficiencia renal con necesidad de hemodiálisis (HD), a pesar de una implementación adecuada de medidas de prevención y tratamiento 1, 2.
El objetivo del presente trabajo es comunicar cuatro casos de SLT con características atípica internados en el Instituto de Investigaciones Médicas Alfredo Lanari, y realizar una revisión de los posibles mecanismos fisiopatológicos involucrados, presentación clínica y tratamiento.
La información de cada caso fue obtenida en forma retrospectiva a través de la revisión de historias clínicas, manteniendo la confidencialidad de los datos.
Caso clínico 1
Varón de 71 años que consultó por disnea clase funcional II, edema de miembros inferiores y aumento del perímetro abdominal de dos meses de evolución. En los resultados de laboratorio se informó anemia normocítica normocrómica, leucocitosis y creatininemia normal con hiperfosfatemia, hiperuricemia y aumento de la LDH. Mediante ecografía se observó ascitis libre de moderada cantidad y una formación sólida, fija al peritoneo parietal supra umbilical.
El líquido ascítico presentó gradiente de albúmina seroascítico > 1.1, lactato deshidrogenasa (LDH) 18560 UI/l, y 256 500 células nucleadas por mm3 con células de aspecto linfoideo, compatible con proceso linfoproliferativo. El inmunofenotipo por citometría de flujo de las células del líquido ascítico apoyó el diagnóstico de linfoma no Hodgkin de células B maduras, particularmente linfoma Burkitt.
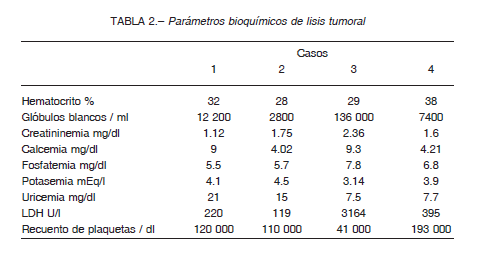
Al ingreso ya presentaba diagnóstico de SLT de laboratorio, por lo que se inició tratamiento con alopurinol 600 mg/día e hidratación parenteral. A las 48 horas agregó insuficiencia renal aguda (Tabla 2) con requerimiento de HD. Al tercer día de internación evolucionó con paro cardiorrespiratorio con ritmo de fibrilación ventricular, sin respuesta a las medidas de reanimación avanzada.
Caso clínico 2
Mujer de 56 años que se internó refiriendo intensos dolores óseos. Al ingreso se constató tricitopenia y una banda monoclonal IgG Lambda en suero con cadenas livianas Lambda en orina. Mediante una biopsia de médula ósea se evidenciaron hallazgos compatibles con mieloma múltiple y amiloidosis. Se indicó hidratación endovenosa, dexametasona 24mg/día y una dosis de 90 mg de pamidronato parenteral. A las 48 horas desarrolló hiperuricemia, hiperfosfatemia y falla renal (Tabla 2). Se diagnosticó SLT clínico y de laboratorio. Inició tratamiento con alopurinol 100 mg cada 12 h. A los cinco días, con función renal normal, pudo ser dada de alta.
Caso clínico 3
Mujer de 41 años que consultó por registros de febrícula. Al examen físico no eran evidentes visceromegalias. Tenía anemia, plaquetopenia, glóbulos blancos 136 000/mm3 y LDH 3164 U/l. La función renal, y los niveles de uricemia, calcemia, fosfatemia y ionograma plasmático de ingreso eran normales.
Una punción de médula ósea confirmó el diagnóstico de leucemia mieloide aguda M5. En presencia de hiperleucocitosis y elevación significativa de la LDH, se indicaron medidas de prevención de lisis tumoral con hidratación endovenosa y alopurinol (600 mg/día). A las 24 h inició tratamiento citorreductor con hidroxiurea en dosis de 50 mg/kg/día, reduciendo la masa leucocitaria al 50% en 36 horas.
Evolucionó a las 48 horas con hiperfosfatemia, hiperuricemia y deterioro de la función renal (Tabla 2). Requirió hemodiálisis transitoriamente.
Posteriormente inició quimioterapia de inducción “7+3” (citarabina 100/mg m2 y daunorrubicina 60 mg/m2/día). Se otorgó el alta a los 30 días de internación con función renal normal.
Caso clínico 4
Varón de 68 años con diagnóstico de linfoma del manto que se internó para recibir quimioterapia. Presentaba linfadenopatías múltiples, esplenomegalia e infiltración difusa de la médula ósea.
Había recibido el primer ciclo de quimioterapia con esquema R-CHOP (rituximab, ciclofosfamida, clorhidrato de doxorubicina, sulfato de vincristina y prednisona) un mes previo a la internación.
Al examen físico se constató esplenomegalia. El laboratorio de ingreso no evidenciaba alteraciones significativas. Se indicó hidratación parenteral y alopurinol 300 mg/día. Inició quimioterapia con esquema R-DHAP (rituximab, cisplatino y citarabina a altas dosis) asociado a dexametasona 40 mg/día y factor estimulante de colonias (filgrastim en dosis de 5 μg/kg/día). A las 48 horas presentó leucocitosis, hiperfosfatemia, hiperuricemia e insuficiencia renal aguda (Tabla 2). Se aumentó la hidratación parenteral y se suspendió la quimioterapia.
Desarrolló shock séptico con foco infeccioso respiratorio. Requirió noradrenalina a dosis de 3 ug/kg/minuto, asistencia respiratoria mecánica y HD. Falleció a los 7 días de instalado el SLT.
Discusión
En nuestra serie los pacientes presentaban enfermedades oncohematológicas, sin embargo, a diferencia de lo clásicamente descripto, en la mayoría de ellos el SLT no pudo ser asociado a drogas citotóxicas.
El primer caso corresponde a un SLT espontáneo (STLE) como inicio de un linfoma de Burkitt. El SLTE es una entidad poco frecuente, cuyo diagnóstico podría estar infraestimado debido a la ausencia de sospecha clínica 4. En este paciente llamaba la atención la presencia de niveles muy elevados de ácido úrico, con fosfatemia ligeramente por arriba del límite superior normal. Se ha descripto que los pacientes con SLTE suelen cursar con niveles de ácido úrico mayores a 15 mg/dl, sin elevaciones significativas de la fosfatemia 5. Una explicación posible para dicha disociación es que, en ausencia de quimioterapia, las células tumorales serían capaces de reutilizar el fósforo liberado durante la lisis celular para continuar con su replicación. El desenlace fue desfavorable pese a las medidas terapéuticas instauradas, probablemente por tratarse de una enfermedad oncohematológica agresiva con gran masa tumoral.
El segundo caso presentaba dos características atípicas. Una de ellas es que la enfermedad de base era mieloma múltiple (MM) y la segunda el desarrollo de SLT asociado al uso de corticoides sistémicos. El SLT en MM se presenta en cerca del 1% de los pacientes que reciben dosis altas de quimioterapia. Su baja incidencia está dada porque el tumor prolifera de manera lenta y es poco quimiosensible. Los que evolucionan con SLT son aquellos que tienen grandes masas tumorales, elevada LDH, morfología plasmoblástica y cariotipos desfavorables 6.
El SLT asociado a corticoides se describe en una baja proporción de casos (6%) 6. La dosis informada para su desarrollo es muy variada, desde dosis moderadas de hidrocortisona y prednisona (200mg y 35mg respectivamente), a dosis altas de dexametasona y metilprednisolona (20 mg y 1g respectivamente). El tiempo descripto para el SLT inducido por corticoides es de 12 a 72 horas luego de su administración6. Si bien se postula que estaría dado por un comportamiento quimio-sensible de las células hematológicas malignas a los corticoides, el mecanismo no está completamente dilucidado.
La mayoría de los casos de SLT en MM han sido comunicados en asociación a esquemas terapéuticos con bortezomib o talidomida 7. En relación a MM y SLT por corticoides, encontramos un único caso con diagnóstico de MM que desarrolló SLT luego de haber recibido solo corticoides sistémicos (40 mg de dexametasona/día durante cuatro días) 8.
El tercer caso resulta de interés por haberse desencadenado tras el inicio de hidroxiurea, situación descripta en la literatura en forma aislada en casos de leucemia mielomonocítica crónica 9, en la transformación leucémica de la policitemia vera 10, y en la leucemia mieloide aguda (LMA)11. En LMA, la hidroxiurea se utiliza para reducir la masa leucocitaria en casos con hiperleucocitosis y evitar complicaciones como la leucoestasis, la coagulación intravascular diseminada y al mismo SLT 11.
Se desconoce por qué algunos pacientes evolucionan con lisis tumoral con hidroxiurea y otros no. Seki y col. plantean que, si bien es conocido que la hidroxiurea actúa principalmente inhibiendo la enzima ribonucleotidasa reductasa, a dosis mayores a 20-30 mg/kg/día podría tener un efecto lítico celular directo 12.
Por último, el cuarto caso también resulta remarcable debido a que el paciente tenía diagnóstico de linfoma del manto; clasificado según la Organización Mundial de la Salud como un linfoma indolente, con bajo riesgo de desarrollar SLT 13. En la literatura hay publicados tres casos de SLT en linfoma del manto, uno de presentación espontánea y otros dos secundarios a ibrutinib 13-15. En nuestro caso identificamos a la esplenomegalia y la infiltración de médula ósea como factores de riesgo de desarrollo de SLT. Muy probablemente el agregado del shock séptico influyó en el desarrollo de la falla renal grave y al desenlace fatal.
En todos nuestros pacientes se llevaron a cabo las medidas generales recomendadas para prevención y/o tratamiento del SLT, a excepción del uso de rasburicase, que no está disponible en nuestro medio. Si bien es conocido que esta droga es mucho más rápida y efectiva que el alopurinol para descender el ácido úrico, no hay estudios concluyentes que demuestren menor mortalidad y menor incidencia de daño renal en relación a su uso 16.
La totalidad de los casos desarrollaron insuficiencia renal aguda, dos presentaron falla renal grave con necesidad de HD de urgencia y fallecieron. La falla renal es la manifestación clínica más frecuentemente descripta 2, 3. El reconocimiento de los factores de riesgo de SLT es fundamental para poder iniciar las medidas de prevención en forma precoz y así poder evitar la insuficiencia renal grave. Tradicionalmente se ha considerado a la hiperuricemia como la causa principal de daño renal en el SLT; sin embargo, la hiperfosfatemia ha ido ganado relevancia desde el uso de rasburicase 16. El uso del rasburicase ha contribuido al registro en aumento en la literatura internacional del número de casos de SLT normouricémicos 16. En este contexto es que, además de las indicaciones clásicas de HD de urgencia, algunos expertos proponen que la sola presencia de valores extremadamente elevados de fosfatemia debe ser considerada indicación de HD precoz 17.
Por último, cabe destacar que la definición clásica de Cairo y Bishop solo incluye a los casos de SLT asociados a quimioterapéuticos, por lo que se requiere del conocimiento de esta entidad para poder sospechar y diagnosticar al SLTE o al asociado a tratamientos no citotóxicos.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Mirrakhimov A, Voore P, Khan M, Ali A. Tumor lysis syndrome: a clinical review. World J Crit Care Med. 2015; 4: 130-8.
2. Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Eng J Med 2011; 364: 1844-54.
3. Rodriguez-Reimundes E, Perazzo F, Vilches A. Síndrome de lisis tumoral en un paciente con cáncer de riñón tratado con sunitinib. Medicina (B Aires) 2011; 71: 158-60.
4. Kekre N, Djordjevic B, Touchie C. Spontaneous tumour lysis syndrome. CMAJ 2012; 184: 913-6.
5. Agnani S, Gupta R, Atray Nk, Vachharajani TJ.D Marked hyperuricemia with acute renal failure: need to consider occult malignancy and spontaneous tumour lysis syndrome. Int J Clin Pract 2006; 60: 364-6.
6. Yang SS, Chau T, Dai MS, Lin SH. Steroid-induced tumor lysis syndrome in a patient with preleukemia. Clin Nephrol 2003; 59: 201-5.
7. Sezer O, Vesole DH, Singhal S, et al. Bortezomib-induced tumor lysis syndrome in multiple myeloma. Clin Lymphoma Myeloma 2006; 7: 233-5.
8. Van de Kerkhof JJ,Peters WG, Visser J, Creemers GJ. Acute tumor lysis syndrome in a patient with multiple myeloma treated with dexamethasone monotherapy. Neth J Me 2001; 59: 83-5.
9. Otrock ZK, Taher AT, Mahfouz RA, Makarem JA, Shamseddine AI. Acute tumor lysis syndrome secondary to hydroxycarbamide in chronic myelomonocytic leukemia. Am J Hematol 2006; 81: 220-1.
10. Ellis AK, Lee DH. Tumor lysis syndrome induced by hydroxyurea therapy for leukemic transformation of polycythemia vera. Am J Hematol 2002; 71: 237-8.
11. Röllig C, Ehninger G. How I treat hyperleukocytosis in acute myeloid leukemia. Blood 2015; 125: 3246-52.
12. Seki JT, Al-Omar HM, Amato D, Sutton DM. Acute tumor lysis syndrome secondary to hydroxyurea in acute myeloid leukemia. Ann Pharmacother 2003; 37: 675-8.
13. Yun S, Vincelette ND, Phan T, Anwer F. Spontaneous tumour lysis syndrome associated with contrast dye iohexol use in mantle cell lymphoma. BMJ Case Rep 2014; 2014. pii: bcr2014204113.
14. Kaur V, Swami A. Ibrutinib-associated tumor lysis syndrome in a patient with mantle cell lymphoma: A case report. J Oncol Pharm Pract 2017; 23: 235-9.
15. Titus-Rains KS, Brown JN, Hammond JM. Irutinib-associated tumor lysis syndrome in chronic lymphocytic leukemia/small lymphocytic lymphoma and mantle cell lymphoma: A case series and review fo the literature. J Oncol Pharm Pract 2018; 24: 544-9.
16. Dinnel J, Moore B, Skiver BM, Pose P. Rasburicase in the management of tumor lysis: an evidence-based review of its place in therapy. Core Evid 2015; 10: 23-38.
17. El-Husseini A, Sabucedo A, Lamarche J, Courville C, Peguero A. Acute kidney injury associated with tumor lysis syndrome: a paradigm shift. Am J Emerg Med 2012; 30: 390.