
SILVIA M. VARAS 1, MARÍA VERÓNICA PÉREZ CHACA 2, NIDIA N. GÓMEZ 2, 3
1 Química Biológica Patológica, Área de Química Biológica, Facultad de Química Bioquímica y Farmacia, Universidad Nacional de San Luis, 2 Laboratorio de Morfofisiología, Área de Morfología, Facultad de Química Bioquímica y Farmacia, Universidad Nacional de San Luis, 3 Instituto Multidisciplinario de Investigaciones Biológicas (IMIBIO-SL), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), San Luis, Argentina
Resumen Los canales de cloruros, de sodio, de bicarbonato y los de agua (aquaporinas) se coordinan para mantener la cubierta líquido superficial de las vías respiratorias, que es necesaria para el aclaramiento mucociliar. El mecanismo general para el transporte de electrolitos y agua depende principalmente de la expresión diferencial y distribución de los transportadores y bombas de iones. Los iones y el agua se mueven a través de las vía paracelular o transcelular. La ruta transcelular del transporte de electrolitos requiere un transporte activo (dependiente de ATP) o pasivo (siguiendo gradientes electroquímicos) de iones. La ruta paracelular es un proceso pasivo que está controlado, en última instancia, por los gradientes electroquímicos transepiteliales predominantes. La fibrosis quística es una enfermedad hereditaria que se produce por mutaciones en el gen que codifica la proteína reguladora de la conductibilidad transmembrana de la fibrosis quística (CFTR) que actúa como un canal de cloro y cumple funciones de hidratación del líquido periciliar y mantenimiento del pH luminal. La disfunción del canal de cloro en el epitelio respiratorio determina una alteración en las secreciones bronquiales, con aumento de su viscosidad y alteración de la depuración mucociliar y que asociado a procesos infecciosos puede conducir a daño pulmonar irreversible. La disfunción del CFTR, también se ha visto implicado en la patogénesis de la pancreatitis aguda, en la enfermedad pulmonar obstructiva crónica y la hiperreactividad en el asma. Existen fármacos que aprovechan los mecanismos fisiológicos en el transporte de iones, con un objetivo terapéutico.
Palabras clave: aclaramiento mucociliar, líquido superficial, fibrosis quística, enfermedad pulmonar obstructiva crónica
Abstract Ion transporters in the lungs. Use as therapeutic targets. The chloride channels, sodium and bicarbonate channels, and aquaporin water channels are coordinated to maintain the airway surface liquid that is necessary for mucociliary clearance. The general mechanism for the transport of electrolytes and fluids depends mainly on the differential expression and distribution of ion transporters and pumps. Ions and water move through the paracellular or transcellular pathways. The transcellular route of electrolyte transport requires an active transport (dependent on ATP) or passive (following electrochemical gradients) of ions. The paracellular pathway is a passive process that is ultimately controlled by the predominant transepithelial electrochemical gradients. Cystic fibrosis is a hereditary disease that is produced by mutations in the gene that encode cystic fibrosis transmembrane conductance regulatory protein (CFTR) that acts as a chloride channel and performs functions of hydration of periciliary fluid and maintenance of luminal pH. The dysfunction of the chlorine channel in the respiratory epithelium determines an alteration in the bronchial secretions, with an increase in its viscosity and alteration of the mucociliary clearance and that associated with infectious processes can lead to irreversible lung damage. CFTR dysfunction has also been implicated in the pathogenesis of acute pancreatitis, chronic obstructive pulmonary disease, and bronchial hyperreactivity in asthma. There are drugs that exploit physiological mechanisms in the transport of ions with a therapeutic objective.
Key words: mucociliary clearance, airway surface liquid (ASL), cystic fibrosis, chronic obstructive pulmonary disease
Dirección postal: Nidia N. Gómez, Ejército de los Andes 905, 5700 San Luis, Argentina
e-mail: gomez.nidia@gmail.com
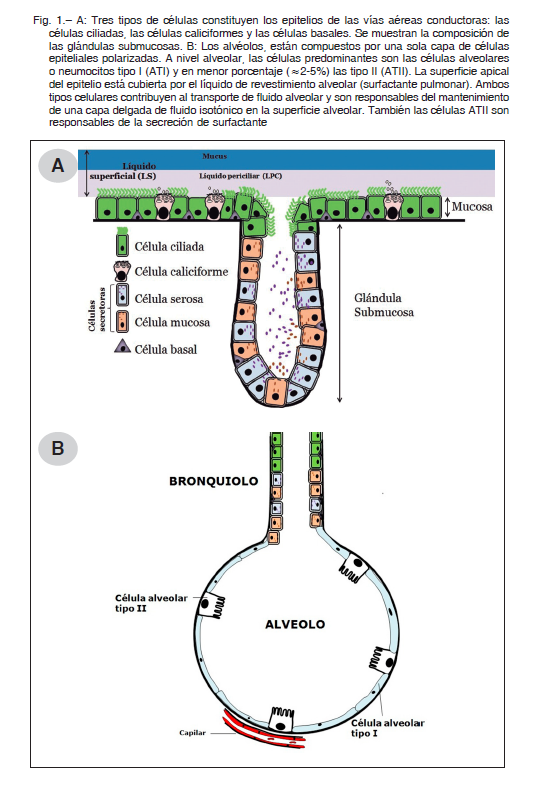
El aparato respiratorio de los mamíferos está constituido por la región alveolar y las vías aéreas. Estas últimas se dividen en una parte de conducción, una parte respiratoria y, finalmente, los alvéolos. El epitelio bronquial está compuesto principalmente de células ciliadas, caliciformes y basales (Fig. 1A), mientras que el epitelio alveolar presenta células alveolares o neumocitos tipo I y neumocitos tipo II (Fig. 1B). El aparato mucociliar está conformado por el epitelio ciliado con células caliciformes, junto con las glándulas submucosas, y tienen como principal función la de atrapar y expulsar las partículas en suspensión que se inhalaron. Las células epiteliales que recubren las vías respiratorias están protegidas del aire inspirado por el líquido superficial (LS) (~10 μm) que se compone de dos capas: una capa de líquido periciliar (LPC) inferior (~7 μm), en donde los cilios se mueven para eliminar las partículas inhaladas y los patógenos atrapados y la capa superior de moco 1. La hidratación del LS es crucial para una eliminación mucociliar eficiente y está estrictamente regulada por el transporte de iones y agua a través de los epitelios y por la secreción de fluidos de las glándulas submucosas 2. La alteración en algún transportador de iones y el subsecuente movimiento de agua constituyen la etiopatogenia de algunas enfermedades de origen hereditario o adquirido como la fibrosis quística (FQ) o la enfermedad pulmonar obstructiva crónica (EPOC).
Ambas enfermedades se caracterizan por el flujo de aire limitado y la pérdida progresiva de la función pulmonar 2. Los pacientes con FQ padecen infecciones bacterianas crónicas e inflamación neutrofílica persistente de las vías respiratorias. La EPOC implica una alteración en el flujo de aire, inflamación y desequilibrio de proteasas 3, 2/3 de los pacientes presentan bronquitis crónica con producción de moco y tos recurrente. Hay casos en que la manifestación principal de la EPOC es el enfisema. Si bien los patógenos bacterianos que se encuentran en los pulmones son diferentes en los pacientes con FQ que, en aquellos con EPOC, la obstrucción del moco de la vía respiratoria inferior está presente en ambas enfermedades, lo que contribuye a la pérdida de la función pulmonar 3, 4.
Existen numerosos trabajos que analizan la manipulación farmacológica de los canales iónicos en pulmón,
para su uso terapéutico en distintas enfermedades 2, 4-7. Por ejemplo; ivacaftor, un medicamento activador del CFTR, usado en pacientes con FQ, cuando es administrado en enfermos con EPOC, recupera eficazmente el defecto funcional del CFTR inducido por el humo del cigarrillo, mejora parcialmente el volumen el líquido superficial de las vías respiratorias y el aclaramiento mucociliar de su epitelio 4, 8-10.
En la presente revisión analizaremos todos los protagonistas del mantenimiento del barrido mucociliar efectivo, como así también los mecanismos responsables de su funcionamiento y algunos ejemplos de su uso como dianas terapéuticas en el tratamiento de enfermedades del pulmón.
Protagonistas responsables del aclaramiento mucociliar
Un aclaramiento mucociliar normal es responsable de limpiar las vías respiratorias de patógenos y partículas inhaladas. Su función depende principalmente de dos parámetros: el barrido mucociliar y el transporte iónico realizado por el conjunto de canales iónicos que expresan sus células. La absorción y secreción iónica permite al epitelio respiratorio controlar el flujo de agua transepitelial y regular la composición del líquido periciliar (LPC), para un barrido ciliar óptimo 1. En condiciones normales, la barrera mucosa funciona como parte del sistema inmune innato; sin embargo, en un cuadro inflamatorio, la producción de moco se acelera como parte de la respuesta del cuerpo a la infección u otras injurias 1, 9.
El moco y la mucina se producen en exceso en pacientes con enfermedades crónicas de las vías respiratorias,
lo que contribuye a la obstrucción de las vías respiratorias en personas con asma, EPOC y FQ 11, 12.
Barrido mucociliar
El barrido mucociliar requiere de tres componentes: la capa de moco, el líquido periciliar y el movimiento de iones en las células epiteliales 12.
Mucinas
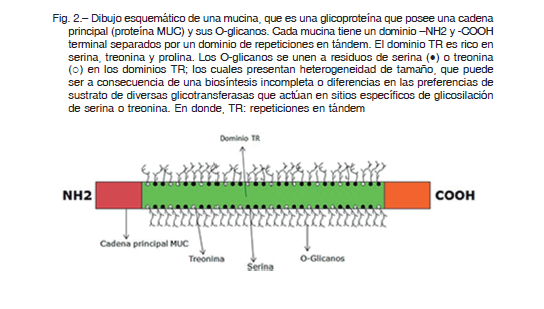
El moco proporciona una barrera fisiológica a toxinas y patógenos ambientales y forma parte de la primera línea de defensa, como parte de la inmunidad innata de la vía aérea. Las mucinas (MUCs) son el principal componente del mucus de la vía aérea y son sintetizadas en las células epiteliales 13. Las mismas son glicoproteínas complejas con un peso molecular de (2-20 × 105 Da), con alto contenido de carbohidratos (50-90%) lo que refleja una gran cantidad de O-glicanos y numerosas repeticiones en tándem (TR) en la cadena principal de la proteína (Fig. 2). Los dominios TR son la característica que distingue a las mucinas de otras glicoproteínas. Las mucinas se clasifican en función de la cadena principal de la proteína MUC y están codificadas por un gen MUC. Los 21 genes que codifican las MUC están localizados en los cromosomas 1, 3, 4, 6, 7, 11, 12 y 1913, de los cuales 14 de ellos se encuentran en el conducto respiratorio 13, 14.
Hay tres clases de mucinas en las vías respiratorias: las que se secretan pero no polimerizan (MUC7), las que se secretan y polimerizan para formar geles (MUC5AC, MUC5B) y las que tienen dominios transmembrana y
están asociadas a la superficie celular (MUC1, MUC4, MUC16 y MUC20) 13. Las dos mucinas principales de la vía aérea son MUC5AC y MUC5B.
Cada TR contiene prolina y es rica en residuos de serina y/o treonina que serán O-glicosilados. Por lo tanto, los TR determinan en gran medida el grado de glicosilación de una mucina, ya que las secuencias TR más largas o más repetidas pueden permitir cientos de O-glicanos por molécula. Así, MUC5B contiene una repetición de 29 aminoácidos con un total de 16 serinas y treoninas. Setenta y dos repeticiones de la secuencia de 29 aminoácidos contribuyen con 1152 posibles sitios de O-glicosilación de la cadena principal de MUC5B1, 13-15 (Fig. 2).
El número de repeticiones del dominio TR en una proteína MUC también determina el tamaño de la mucina, ya
que a medida que se alarga la cadena principal de la proteína aumenta la masa molecular de la misma. También, es probable que contribuyan a la diversidad estructural y funcional de las mucinas en estructuras, aún no bien conocidas, a nivel molecular o fisiológico.
Las mucinas se localizan en células específicas, así MUC5AC se encuentra en las células caliciformes de las vías respiratorias. MUC5B se encuentra en las células mucosas de la glándula submucosa y en el epitelio, MUC7 se ubica en las células serosas de la glándula submucosa, MUC16 está asociado con el dominio apical de las células caliciformes de las vías respiratorias y MUC1, MUC4 y MUC20 están localizados en células ciliadas de la vía aérea 13, 15. Se sabe que MUC5B y MUC5AC forman redes diferentes, en donde MUC5B tiende a formar grandes polímeros lineales, mientras que MUC5AC forma estructuras similares a redes 6. Por otro lado, las células serosas son el origen primario del componente fluido de las secreciones y de las moléculas de la inmunidad innata. En pacientes con diversas enfermedades pulmonares obstructivas como EPOC y en el asma se ha visto hiperplasia de las células caliciformes y una expresión alterada del ARNm de MUC5B, mientras que en pacientes con FQ solo hay
hipertrofia de estas células 13-18 a pesar de presentar un moco deshidratado anormal14, 18 que es mejorado con la terapia moduladora sobre CFTR 2.
Transportadores responsables de la absorción y secreción de iones
Los principales canales responsables del movimiento de iones son los canales CFTR, los canales epiteliales de sodio (ENaCs), la familia de transportadores de solutos (SLC), los canales de cloruros dependientes de calcio (ANO1) y los canales de cloruros ClC. El funcionamiento de los canales, además está asociado al movimiento de agua por medio de acuaporinas.
Regulador transmembrana de la fibrosis quística
El CFTR es una glicoproteína compuesta por 1480 aminoácidos y un peso molecular de 180 KDa. Tiene 5
dominios, 2 de ellos son transmembrana (TM), cada uno con seis pasos a través de membrana, los cuales se unen a los dos dominio de unión a nucleótido citoplasmático (NBD1 y 2). El NBD1 está a su vez conectado a un sitio regulatorio R 19. El CFTR es activado por fosforilación de su dominio regulatorio intracelular R, por la proteína quinasa dependiente de AMPc (PKA) que es activada por la forskolina (un activador de la adenilato ciclasa). Una vez que el canal es activado por la fosforilación, las aperturas y cierres son regulados por hidrólisis del ATP en los dominios TM1 y TM2 y por el ATP intracelular que se une a los sitios NBD, los cuales son inactivados por fosfatasas 19.
La expresión y función de CFTR se ha descripto en el epitelio de la vía respiratoria superior e inferior. La mayoría de los estudios de localización identifican al CFTR en la membrana plasmática apical de células ciliadas en el epitelio nasal y de las vías respiratorias y las células serosas acinares de las glándulas submucosas de los bronquios y más recientemente se los ha identificado, en células alveolares tipo II. CFTR regula otros canales iónicos y transportadores y está críticamente involucrado en la regulación del pH de LS y en la inmunidad innata, esto último mediante la regulación de la secreción de moléculas antioxidantes 20 y antimicrobianas 17.
Canales epiteliales de sodio
Los canales epiteliales de sodio (ENaCs) son una subfamilia de canales iónicos dentro de la superfamilia Degenerin / ENaC (DEG / ENaC. Estos canales iónicos se encuentran en diferentes epitelios que incorporan sodio, incluido el epitelio del colon, el de las vías respiratorias y en los túbulos en las nefronas; su actividad representa el paso limitante de la absorción de sodio y por lo tanto, del movimiento del agua transepitelial. La captación de sodio en estos epitelios ocurre a través de un mecanismo reconocido como la “hipótesis de las dos membranas”, donde el sodio ingresa a la cara apical de la célula epitelial a través de ENaCs para luego ser expulsado por la cara basolateral por la bomba Na+/K+-ATPasa. Esta absorción electrogénica de Na+ crea un gradiente osmótico transepitelial que representa la principal fuerza motriz del movimiento del agua a través del epitelio.
El complejo multiproteico de ENaC consta de tres subunidades homólogas (α, β y γ) y se ha sugerido que el canal iónico se ensambla con una estequiometria de 2α:1β:1γ. Las ENaC están formadas por subunidades
cuyos extremos incluyen -NH2 y -COOH intracelulares, dos dominios transmembrana y una gran asa extracelular glicosilada con dominios ricos en cisteína 21. La proteína SPLUN1 (del inglés secreted protein, short palate lung and nasal epitelial clone1) se encuentra presente en el LS y actúa regulando el volumen de este líquido uniéndose al ENaC y previniendo la escisión y activación por proteasas; esta acción es inhibida cuando el LS presenta pH ácido 2.
La actividad desregulada de ENaCs contribuye a la patogénesis de enfermedades humanas que están asociadas
con una homeostasis alterada de sodio y/o agua.
En la vía respiratoria, la hiperactividad de ENaC puede conducir al agotamiento de la capa de LS del epitelio y dar como resultado una enfermedad pulmonar similar a la FQ.
Por el contrario, la hipoactividad de los ENaCs contribuye al desarrollo de edema pulmonar debido a la
alteración del aclaramiento del líquido alveolar.
El aumento de la acumulación de fluido puede resultar de una función de ENaC comprometida cuando:
– Una fuerza hidrostática asimétrica de la vasculatura empuja el fluido desde los capilares hacia el espacio alveolar por ejemplo por hipertensión pulmonar o por alteraciones de las uniones estrechas (tight junction), lo que da como resultado un aumento de la permeabilidad.
– Durante la lesión pulmonar aguda (sepsis, inflamación o infección) se producen citoquinas inflamatorias que inhiben los canales ENaC y causan cambios en el transporte de Na+. Una disminución de la reabsorción de sodio por estos canales permite que el líquido se acumule a nivel alveolar, con la consecuente pérdida del intercambio normal de gases y formación de edema e hipoxemia 22-24.
Familia de transportadores de soluto
La gran familia de transportadores de soluto (SLC) comprende numerosos grupos de integrantes. Mencionaremos solo los descriptos en pulmón: integrantes de la familia SLC4, SLC12 y SLC26 (ver Tabla 1).
Familia SLC4: familia de transportadores de bicarbonato
NBC: son co-transportadores de NaHCO3 -, de los cuales existen dos isoformas: pNBC1, también conocidos
como SLC4A4, NBCe1 y NBC4 llamados como SLC4A5, NBCe2. Estos transportadores se expresan en la membrana
basolateral 25. Los inhibidores de la secreción de HCO3 – son acetazolamida (inhibidor de la anhidrasa carbónica) y DIDS (inhibidor de los intercambiadores Cl- / HCO3 -).
AE: es un intercambiador aniónico ubicado en la membrana basolateral, cuya función es ingresar Cl– y excretar HCO3 -, pertenece a la familia SLC4A2. El canal AE2 trabaja en paralelo con el co-transportador Na+-bicarbonato (NBC). De esa forma, es una vía de acumulación de Cl-, en células epiteliales 26.
Familia SLC12: familia de co-transportadores de cloruros acoplados a catión electroneutro
Cotransportador Na+-K+-2Cl- (NKCC) es una proteína transmembrana que facilita el co-transporte de un ion sodio, uno de potasio y dos de cloro desde el espacio extracelular hacia el interior de las células. La isoforma NKCC1 es codificada por SLC12A2 y el NKCC2 por SLC12A1. Estos transportadores son inhibidos por bumetanida, furosemida y otros compuestos estructuralmente similares. Además, se ha informado que los bloqueadores de NKCC1 pueden inhibir el crecimiento de células cancerosas 12.
Familia SLC26: familia de intercambiadores aniónicos multifuncionales
Pendrin (o SLC26A4): es un intercambiador de aniones transmembrana que se encuentra en la superficie apical de las células epiteliales y media el intercambio Cl-/HCO3-. Su expresión es fuertemente inducida por IL-4, IL13 e IL-17A. En estudios recientes pendrin fue identificado como el gen más altamente regulado en bronquios de pacientes asmáticos y se corroboró su expresión en modelos murinos de EPOC 27-30.
SLC26A9: es un canal ubicado en la membrana apical en donde tiene funciones como intercambiador Cl-/HCO3
-, además como canal de cloruros y como co-transportador de sodio-anión 12.
ANO1 o ANOCTAMIN 1 (o TMEM16A): es un canal de cloruro activado por calcio, identificado en el año 2008.
TMEM16A y sus parálogos han sido investigados activamente por múltiples funciones para controlar el flujo de cloruros (activados por Ca2+), en una variedad de células epiteliales secretoras. Además, TMEM16A es estimulado por agonistas purinérgicos. La estimulación de los receptores purinérgicos luminales causa un aumento del AMPc y calcio intracelular 31-33. El AMPc induce la secreción de Cl- a través de la activación de CFTR en el lado luminal, mientras que el Ca2+ intracelular provoca la secreción de Cl-, a través de la activación de TMEM16A. Mientras que CFTR se expresa predominantemente en células ciliadas, TMEM 16A es más abundante en células caliciformes secretoras de moco. Ambos canales de cloruros se expresan en las glándulas submucosas de los bronquios. Los niveles de transcrito TMEM16A son de aproximadamente 10 a 100 veces más abundantes que los de CFTR en las células serosas y mucosas, respectivamente. Los ratones KO para TMEM16A mueren en pocas semanas después del nacimiento, principalmente con traqueomalacia, que es una malformación grave de la tráquea. En los ratones KO se ha observado la acumulación grave de moco en el lumen de la tráquea, lo que sugiere una hidratación inadecuada en las vías respiratorias.
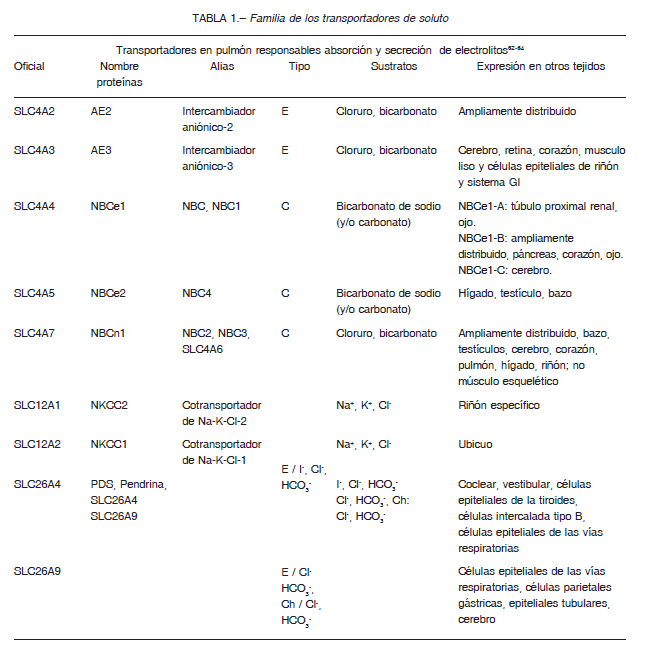
Estos canales de Cl- transportan otros aniones como bicarbonato, a través de la membrana apical hacia el LS que cubre las vías respiratorias.
Los canales CFTR están controlados por la vía de señalización del AMPc. Los canales de Cl- dependiente de
Ca2+ se abren cuando la concentración de Ca2+ citosólico aumenta y media la actividad secretora de los cloruros, en respuesta a la estimulación purinérgica 34-39. Así, los receptores purinérgicos, P2Y2 se expresan tanto en las células ciliadas como en las células caliciformes, mientras que los receptores A2b solo se encuentran en las células ciliadas. En LS la concentración de los ligandos de estos receptores, ATP y adenosina (A), están controlados por un conjunto complejo de enzimas metabolizadoras de nucleótidos que incluyen ectonucleotidasas, fosfatasas no específicas y con actividades transfosforilantes. El ATP liberado es rápidamente hidrolizado, pero la remoción de adenosina es 10 veces más lenta que la del ATP; lo que resulta en acumulación de adenosina en el epitelio. La eliminación de las moléculas de ATP del espacio extracelular se produce por conversión a adenosina por enzimas extracelulares solubles (CD39 y CD73). La adenosina vuelve al citoplasma a través de transportadores específicos ENT1, ENT2 (del inglés, equilibrative nucleoside transporter) y también CNT1, CNT2 (CNT: concentrative nucleoside transporters).
La activación de receptores A2b por adenosina causa la activación de proteína Gs activa la enzima adenilato
ciclasa (AC) que convierte al ATP citosólico en el segundo mensajero AMP cíclico (AMPc). La interacción ATP- R purinérgico P2Y2, interactúa con la proteína Gq, se forma el complejo Gqα-GTP y se activa la fosfolipasa C (FLC) de membrana. FLC hidroliza el fosfatidil-inositol 4,5- bisfosfato (PIP2) a IP3 + diacilglicerol (DAG). IP3 estimula la liberación de Ca2+ y DAG a PKC (protein kinasa C).
Las dos vías de secreción de cloruros por ANO1 y CFTR tendrían un cross-talk 40 (Figura 3).
La expresión epitelial de TMEM16A aumenta en los asmáticos y en condiciones proinflamatorias, por ejemplo
en el tratamiento con citoquinas o componentes bacterianos 41.
Canales de cloruros
La regulación del volumen celular es esencial para cada célula y se logra mediante la pérdida o ganancia regulada de iones intracelulares. La disminución del volumen a menudo implica la activación paralela de los canales de potasio y cloruro. Los canales de cloruro (ClCs) son permeables para iones cloruro u otros iones en la membrana plasmática. Las proteínas ClC están codificadas por genes de la familia ClC. La familia de estos canales se clasifican según su ubicación en las células en dos subfamilias distintas: ClC1, ClC2 y ClC-Ka/Kb-canales Barttin que se ubican en todas las membranas plasmáticas y ClC3, ClC4, ClC5, ClC6 y ClC7 en membranas de endosomas y lisosomas 42-43.
ClC2 atrajo una creciente atención ya que se pensaba que la misma posee diversas propiedades moleculares,
funcionales y fisiológicas. Este canal se lo asoció con varias enfermedades, incluido el cáncer de pulmón. Se ha demostrado que ClC2 afecta la producción de líquido pulmonar y la morfología del quiste pulmonar 42-44. Algunos autores propusieron que era capaz de compensar la pérdida de los canales CFTR, en la FQ. Esta hipótesis, sin embargo, requiere que ClC2 esté presente en las membranas apicales, una propuesta que se considera incorrecta 42. La localización de ClC2 en la parte apical o basolateral de las células epiteliales fue motivo de gran controversia. Actualmente, está firmemente establecido que ClC2 se expresa en la membrana basolateral y en los complejos de uniones estrechas de los epitelios. Este tipo de canal puede activarse por hiperpolarización, aumento del volumen celular, hipotonicidad y acidificación extracelular. Adicionalmente, se ha informado que ClC2 es débilmente sensible a los clásicos bloqueadores como DID o 9-AC, como también al Zn2+ y Cd2+ y es regulado por hormonas (tiroideas y estrógenos), drogas, proteínas, quinasas, colesterol de las membranas y distintos factores de transcripción 45-47.
Acuaporinas
El transporte de fluidos sigue algunos principios como el gradiente osmótico, el mecanismo de Starling (debido a la presión hidrostática) y la pinocitosis. Hay 4 acuaporinas (AQP) que se expresan en los pulmones, ellas son AQP1 (en capilares), AQP3, AQP4 y AQP5. Se sabe que tanto AQP1, como AQP5 principalmente facilitan el transporte de fluido osmótico a través de la membrana apical de las células endoteliales y las células epiteliales alveolares; pero no pueden participar en el transporte de fluidos impulsados por permeabilidad capilar y por los cambios de presión hidrostática. AQP4 se expresa en la membrana basolateral de los epitelios bronquiales, de la tráquea y en células epiteliales alveolares en ratas 48. AQP5 se expresa en las membrana apicales de las células alveolares tipo I en humanos y en las células alveolares tipo II (solo en ratones) y en las células serosas de las glándulas submucosas de la vía aérea superior 11.
En las glándulas submucosas al estimular las células serosas, a través de los receptores muscarínicos, se
observa un incremento de AMPc citosólico y activación CFTR que induce la secreción de cloruro y de sodio en la célula, para poder mantener la electroneutralidad, a través de la vía intracelular y paracelular y luego el agua sale de las células siguiendo el gradiente osmótico, principalmente a través del canal de agua AQP5.
La eliminación de AQP5 reduce significativamente la secreción de fluido de la glándula y hace que el fluido secretado sea más viscoso 49. Por lo que es interesante probar si la modulación de AQP5 podría ser útil para promover el aclaramiento de moco, de las vías respiratorias en pacientes con EPOC o bronquiectasias. Varios estudios demostraron que AQP1 y AQP5 disminuyen significativamente, después de la lesión pulmonar 49.
Todavía no está claro el papel exacto de los AQP en las enfermedades pulmonares, por lo que se necesita
un mayor número de estudios para dilucidar los roles de las AQP en los procesos fisiológicos y fisiopatológicos del pulmón.
Sistema de absorción y secreción de iones en la célula epitelial
El tejido epitelial se compone de una o más capas de células estrechamente ensambladas que tapizan una superficie o cavidad; siendo la polarización una de las principales características de las células epiteliales. Entre las características a destacar de los epitelios es que las membranas apicales y basolaterales tienen papeles distintos debido a su localización y expresión diferencial de proteínas 11, 12, 16 (Fig. 4).
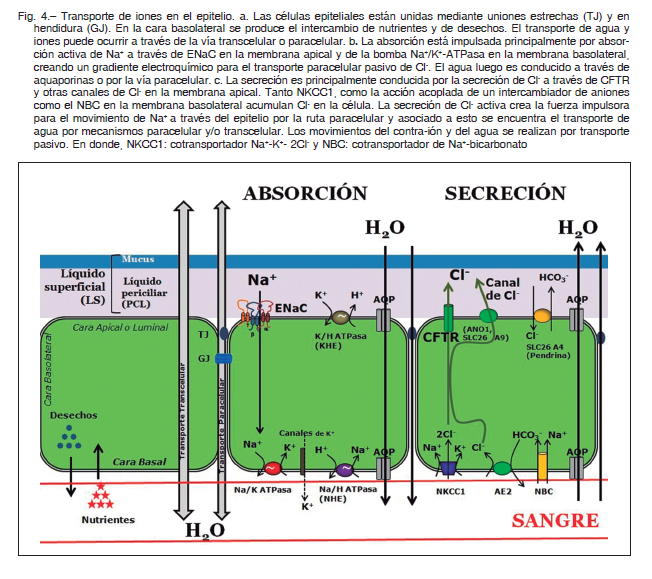
Los sistemas de absorción y secreción de iones son:
1. Reabsorción transepitelial de sodio: se realiza por la actividad concertada de los canales de sodio ENaC de ubicación apical y la bomba Na+/K+ ATPasa que se encuentra basolateral. Los canales de cationes dependientes de nucleótidos cíclicos apicales (CNG) también podrían contribuir a la reabsorción de Na+. Además, un intercambiador Na+/H+ (NHE) ha sido identificado en las células epiteliales de las vías respiratorias para la regulación del pH intracelular.
2. Secreción apical de cloruros: en seres humanos está secreción esta mediada principalmente por canales CFTR y en menor medida por canales de Cl- dependientes de Ca2+ (CACC), tales como los canales TMEM16A (ANO1).
Esta secreción es mantenida por el co-transportador basolateral Na+/ K+/ 2Cl – y el intercambiador aniónico HCO-3 / Cl- (AE). Además, se han identificado tres tipos de canales de cloruros basolaterales: un canal rectificador externo basolateral (basolateral outwardly rectifying Clchannel, BORC), un canal rectificador interno basolateral (basolateral inwardly rectifying Cl- channel, BIRC) y un canal basolateral similar a CFTR (BCFTR). Se ha sugerido que estos canales están implicados en la modulación de la secreción apical de cloruros; además se ha estimado la contribución de cada uno de estos canales en la conductancia de cloruros a nivel basolateral, siendo BORC (86.5%) > BCFTR (8.3%) > BIRC (5.4%).
3. Los canales de K+ han sido identificados en el epitelio de la vía aérea que modulan la secreción apical de Cl-. En la membrana basolateral se han identificado varios canales de K + dependientes de voltaje (Kv7.1-Kv7.5). Por último, en la membrana apical y basolateral se reconoce la presencia de canales de potasio activados por niveles de calcio como SK4, BKCa, KCa 3.1 41, 50-51.
Regulación de la hidratación en células epiteliales: absorción de iones
Las células epiteliales transportan electrolitos que son necesarios para diversos procesos como regulación metabólica, excreción de productos de desecho y protección.
La cantidad total de secreciones se ajusta drásticamente por su velocidad y dirección en respuesta a estímulos externos. El flujo de cloro a través de las células epiteliales secretorias es un determinante importante de la secreción de líquidos y electrolitos.
El co-transportador NKCC1 ubicado en la membrana basolateral de estos epitelios mantiene la concentración
electroquímica de cloruros de hasta cinco veces más a la encontrada en una célula no epitelial. La fuerza de conducción para la captación de Cl- es proporcionada por el gradiente de Na+ que está establecido por la bomba Na+/H +-ATPasa. La alta concentración de Cl- es un factor clave para expulsarlo hacia la luz. Así, el eflujo de Cl- se neutraliza eléctricamente por la descarga de K+ a través de sus canales específicos. Esta acumulación de cargas positivas luminales establece un gradiente osmótico transepitelial para impulsar el movimiento del líquido 11, 12, 50. La absorción de Na+ generalmente se realiza por la activación de canales ENaC. Así, el flujo de Na+ a través de la membrana basolateral es impulsado por la bomba Na+/ K+- ATPasa, que finalmente mantiene un gradiente de Na + (dirigido hacia el citoplasma), necesario para la absorción.
Los canales de K+ tanto apical, como basolateral son esenciales para mantener un potencial de membrana
negativo adecuado para que se produzca la afluencia de Na+ (Figura 4) 11-12.
Secreción de iones
La secreción de iones cloruro y sodio transcelular es controlada principalmente por la salida de Cl- a través de la membrana apical y esto se produce predominantemente a través de CFTR, aunque existen un número de canales de Cl-, tales como los canales de Cl- activados por calcio (CACC) y del transportador SLC 26A9 (intercambiador de HCO3-). La secreción depende del transporte activo de Cl- que crea la fuerza motriz para Na+ (Fig. 4). El aumento de la concentración de NaCl en la superficie apical (o luminal) genera una fuerza motriz osmótica necesaria para que el agua se secrete, produciendo una secreción
isotónica. Para ambos procesos, el agua se mueve de forma pasiva a través de la vía paracelular o transcelularmente por medio de las acuaporinas, o por ambas vías. CFTR y pendrina (SLC26A4) también trasladan HCO-3 que juega un papel clave en la modulación del pH del fluido secretado 51.
Las células epiteliales de las vías respiratorias, así como las células serosas de las glándulas submucosas secretan Cl- y HCO3 – en respuesta a agentes que aumentan el AMPc intracelular (VIP, adenosina y noradrenalina) y / o Ca2+ (acetilcolina, histamina o ATP) y de esa forma se controla el espesor y la composición del líquido periciliar (LPC).
En resumen, la absorción de Na+ por ENaC en células epiteliales y su transporte activo por la Na+/K+-ATPasa producen un gradiente electroquímico transmembrana que permite el cotransporte de Na+, K+ y Cl – a través de NKCC1 en la membrana basolateral. Lo que aumenta hasta cinco veces la concentración del Cl- intracelular, por encima del equilibrio. También, en células serosas se ha demostrado que Cl- se puede acumular en la membrana basolateral, por otros dos mecanismos: (1) a través de la acción acoplada de un intercambiador Na+/H+ (NHE) y un intercambiador Cl-/HCO-3 y (2) por el funcionamiento en paralelo de un intercambiador Cl-/HCO3- con NBC. En la membrana apical, Cl – se secreta a través de canales y transportadores CFTR, ANO1 o SLCs.
Fisiopatología
En los pacientes con FQ el LS es ácido (por el transporte defectuoso de Cl- y bicarbonatos); donde la regulación de ENaC se interrumpe, dando como resultado un incremento de la absorción de sodio y mayor agotamiento del volumen del líquido superficial de la vía aérea. Esta situación genera una deshidratación y adelgazamiento del LS. En estado deshidratado, el agua se mueve preferentemente fuera de la capa de moco, aumentando su concentración. El volumen de líquido periciliar se conserva inicialmente, pero finalmente alcanza un umbral crítico en el que el agua también sale. La capa de moco posteriormente comprime la película de líquido periciliar, comprometiendo la actividad ciliar y el aclaramiento mucociliar 50, 51.
Para el tratamiento de pacientes FQ se usa inhibidores selectivos de ENaC como amilorida, que genera rehidratación LS y aumento de la función ciliar 52. Se ha observado que el bloqueo excesivo de ENaC podría causar un daño grave debido a hipercalemia o acumulación indeseable de líquido en los pulmones (edema pulmonar) 21.
Canales de iones como dianas terapéuticas
La regulación o activación de canales alternativos de cloruro apical, incluyendo el transportador SLC26A4, A9 y anoctamina1 podrían corregir el transporte defectuoso de cloruro apical y por lo tanto las anomalías del LS. Los inhibidores de proteasa y los derivados del SPLUNC1- insensibles al pH podrían resultar beneficiosos para regular la actividad de ENaC, pero se necesitan investigaciones en humanos 41, 52-53.
Se ha demostrado que la inhibición de canales como TMEM16A, que están asociados a la modulación de
la actividad del co-transportador NKCC conducen a la relajación del músculo liso y así junto con los agonistas β proporcionan una nueva base para el tratamiento del asma 17.
Recientemente, un grupo de pacientes con EPOC tratados con ivacaftor, un potenciador del CFTR, tuvieron
mejoras no significativas en la función de CFTR, en comparación con los controles. Las expectativas aumentaron con roflumilast, el cual es un inhibidor de la fosfodiesterasa-4 (PDE-4). Roflumilast activa CFTR al aumentar su fosforilación de una manera competitiva con los estímulos dependientes de AMPc. Su administración aumento la actividad de CFTR, con aumento del espesor de LS 54-58.
Se plantea una asociación sinérgica con la acción combinada de potenciadores asociados a PDE-4 57. Por último, se ha estudiado los efectos de los micro-ARNs sobre la expresión de ENaCs, como estrategia para manipular la expresión de los mismos en pulmón 58.
Adicionalmente, debido a que existe un aumento de la expresión de pendrina en modelos de ratones con asma
o EPOC, con aumento de la expresión de MUC 5AC se postula como un blanco terapéutico potencial para tratamiento de asma y EPOC 59.
Estudios recientes han resaltado la importancia de la regulación del pH del LS. Se ha demostrado un pH
ácido de LS en pacientes con FQ y otras enfermedades pulmonares como asma, EPOC y el síndrome de dificultad respiratoria aguda 1, 60. La causa principal de EPOC es una exposición crónica a injurias oxidativas, como el tabaquismo o la exposición pasiva al humo del cigarrillo.
Se sabía que la exposición al humo del cigarrillo disminuía la secreción de cloruros luego se demostró que este efecto era por una inhibición del CFTR, que conduce a la deshidratación del LS, lo que contribuiría al desarrollo de la EPOC 60.
Nuevas evidencias demuestran la falta de secreción de HCO3- en las vías respiratorias de personas con FQ,
lo que conduce a un pH ácido del LS. Se ha postulado que la pérdida de HCO3- estaría afectando la expansión
de mucina y la formación de gel mucoso 15, 17, 26.
Conclusión y perspectivas
CFTR tiene un papel fundamental en la fisiología de las vías respiratorias. Este canal está involucrado directa e indirectamente en la secreción de aniones cruciales para la hidratación de las vías respiratorias, como también el pH y espesor del LS. Las terapias potenciales para la FQ han demostrado ser difíciles de implementar, posiblemente porque CFTR está involucrado en diferentes funciones, en las vías respiratorias. Se están desarrollando otras formas de compensar los CFTR defectuosos mediante la activación de canales iónicos alternativos o transportadores, tales como los miembros de la familia SLC26 (A4 o A9) o las anoctaminas y asociados como CLCA1 61. Estas terapias podrían ser utilizadas tanto para enfermedades hereditarias, como la FQ, o para enfermedades adquiridas como EPOC.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Ma J, Rubin BJ, Voynow JA. Mucins, Mucus, and Goblet Cells. Chest 2018; 154: 169-76.
2. Haq IJ, Gray MA, Garnett JP, et al. Airway surface liquid homeostasis in cystic fibrosis: pathophysiology and therapeutic targets. Thorax 2016; 71: 284-7.
3. Burney P, Perez-Padilla R, Marks G, et al. Chronic Lower Respiratory Tract Diseases. En: Prabhakaran D, Anand S, Gaziano TA, et al (eds). Cardiovascular, Respiratory, and Related Disorders (3rd ed). Washington (DC): The International Bank for Reconstruction and Development / The World Bank; 2017, Chapter 15. En: https://www.ncbi.nlm.nih.gov/books/NBK525159/; consultado septiembre 2018.
4. Sloane PA, Shastry S, Wilhelm A, et al. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS One 2012; 7: e39809
5. Palma AG, Kotsias BA, Marino GI: Funciones de los canales iónicos CFTR y ENAC en la fibrosis quística. Medicina (B Aires) 2014; 74: 133-9.
6. Martiniano SL, Toprak D, Ong T, et al. Highlights from the 2017 North American Cystic Fibrosis Conference. Pediatric Pulmonol 2018; 53: 979-86.
7. Mall MA, Hartl D. CFTR: cystic fibrosis and beyond. Eur Respir J 2014; 44: 1042-54.
8. Dransfield MT, Wilhelm AM, Flanagan B, et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 2013; 144: 498-506.
9. Raju SV, Jackson PL, Courville CA, et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am J Respir Crit Care Med 2013; 188: 1321-30.
10. Courville CA, Tidwell S, Liu B, et al. Acquired defects in CFTR-dependent beta-adrenergic sweat secretion in chronic obstructive pulmonary disease. Respir Res 2014; 15: 25.
11. Hahn A, Faulhaber J, Srisawang L, et al. Cellular distribution and function of ion channels involved in transport processes in rat tracheal epithelium. Physiol Rep 2017; 5: e13290.
12. Hollenhorst MI, Richter K et al. Ion transport by pulmonary epithelia. J Biomed Biotechnol 2011; 2011: 174306.
13. Jaramillo AM, Azzegagh Z, Tuvim MJ, et al. Aiway mucin secretion. Ann Am Thorac Soc 2018; 15: S164-70.
14. Ehre C, Ridley C, Thornton DT. Cystic fibrosis: an inherited disease affecting mucin-producing organs. Int J Biochem Cell Biol 2014; 52: 136-45.
15. Kreda SM, Davis CW, Callaghan RM. CFTR, mucins, and mucus obstruction in cystic fibrosis. Cold Spring Harb Perspect Med 2012; 2:a009589.
16. Brune K, Frank J, Schwingshack A, et al. Pulmonary epithelial barrier function: some new players and mechanisms. Am J Physiol Lung Cell Mol Physiol 2015; 308: L731-45.
17. Bartoszewski R, Matalon S, Collawn JF. Ion channels of the lung and their role in disease pathogenesis. Am J Physiol Lung Cell Mol Physiol. 2017; 313: L859-72.
18. Ha EV, Rogers DF. Novel therapies to inhibit mucus synthesis and secretion in airway hypersecretory diseases. Pharmacology 2016; 97: 84-100.
19. Elborn JS. Cystic fibrosis. Lancet 2016; 388: 2519-31.
20. Prakash YS, Pabelick CM, Sieck GC. Mitochondrial dysfunction in airway disease. Chest 2017; 152: 618-26.
21. Althaus M. ENaC Inhibitors and airway re-hydration in cystic fibrosis: state of the art. Curr Mol Pharmacol 2013; 6: 3-12.
22. Matthay MA. Resolution of pulmonary edema. Thirty years of progress. Am J Respir Crit Care Med 2014; 189: 1301-8.
23. Fanelli V, Ranieri VM. Mechanisms and clinical consequences of acute lung injury. Ann Am Thorac Soc 2015; 12 Suppl 1: S3-8.
24. Matalon S, Bartoszewski R, Collawn JF. Role of epitelial sodium channels in the regulation of lung fluid homeostasis. Am J Physiol Lung Cell Mol Physiol 2015; 309: L1229-38.
25. Bonar PT, Casey JR. Plasma membrane Cl-/HCO3- exchangers: structure, mechanism and physiology. Channels
(Austin) 2008; 2: 337-45.
26. Kunzelmann K, Schreiber R, Hadorn HB. Bicarbonate in cystic fibrosis. J Cyst Fibros 2017; 16: 653-62.
27. Chun-E Jia, Dingyuan Jiang, Huaping Dai, et al. Pendrin, an anion exchanger on lung epithelial cells, could be a novel target for lipopolysaccharide-induced acute lung injury mice. Am J Transl Res 2016; 8: 981-92.
28. Dossena S, Nofziger C, Lang F, et al. The ESF Meeting on “The Proteomics, Epigenetics and Pharmacogenetics of Pendrin”. Cell Physiol Biochem 2011; 28: 377-84.
29. Nofziger C, Dossena S, Suzuki S, et al. Pendrin function in airway epithelia. Cell Physiol Biochem 2011; 28: 571-8.
30. Kim D, Huang J, Billet A, et al. Pendrin mediates bicarbonate secretion and enhances CFTR function in
airway surface epithelia. Am J Respir Cell Mol Biol 2019; doi: 10.1165/rcmb.2018-0158OC. [Epub ahead of print]
31. Wang J, Haanes KA, Novak I. Purinergic regulation of CFTR and Ca2+-activated Cl− channels and K+ channels in human pancreatic duct epithelium. Am J Physiol Cell Physiol 2013; 304: C673-84.
32. Yongwoo J, Uhtaek OH. Anoctamin 1 in secretory epithelia. Cell Calcium 2014; 55: 355-61.
33. Lazarowski ER, Schwarzbaum PJ. Señales purinergicas. Medicina (B Aires) 2009; 69: 267-76.
34. Burnstock G, Knight GE. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal 2018; 14: 1-18.
35. Hasan D, Blankman P, Nieman GF. Purinergic signalling links mechanical breath profile and alveolar mechanics with the pro-inflammatory innate immune response causing ventilation-induced lung injury. Purinergic Signalling 2017; 13: 363-86.
36. Caruso M, Alamo A, Crisafulli E, et al. Adenosine signaling pathways as potential therapeutic targets in respiratory disease. Expert Opin Ther Targets 2013; 17: 761-72.
37. Burnstock G, Dale N. Purinergic signalling during development and ageing. Purinergic Signalling 2015; 11: 277-305.
38. Davis CW, Lazarowski E. Coupling of Airway Ciliary Activity and Mucin Secretion to Mechanical Stresses by Purinergic Signaling. Respir Physiol Neurobiol 2008; 163: 208-13.
39. Lazarowski ER, Boucher RC. Purinergic receptors in airway epithelia. Curr Opin Pharmacol 2009; 9: 262-7.
40. Lérias J, Pinto M, Benedetto R, et al. Compartmentalized crosstalk of CFTR and TMEM16A (ANO1) through
EPAC1 and ADCY1. Cell Signal 2018; 44: 10-9.
41. Li H, Salomon JJ, Sheppard DN, et al. Bypassing CFTR dysfunction in cystic fibrosis with alternative pathways for anion transport. Curr Opin Pharmacol 2017; 34: 91-7.
42. Jentsch TJ, Pusch M. CLC Chloride channels and transporters: Structure, function, physiology, and Disease. Physiol Rev 2018; 98: 1493-590.
43. Wang H, Xu M, Kong Q, et al. Research and progress on ClC-2. Mol Med Rep 2017; 16: 11-22.
44. Xu ZN, Zheng XY, Zhao W, et al. Expression and significance of chloride channel ClC2 in nonsmallcell lung cancer. Chin J Lab Diagn 20; 1216: 602.
45. Pasantes-Morales H. Channels and volume changes in the life and death of the cell. Mol Pharmacol 2016; 90: 358-70.
46. Felippe Gonçalves-de-Albuquerque C, Ribeiro Silva A, Ignácio da Silva C, et al. Na/K Pump and Beyond: Na/KATPase as a modulator of apoptosis and autophagy. Molecules 2017; 22: E578.
47. Okada Y, Okada T, Sato-Numata K, et al. Volume-activated and volume-correlated anion channels in mammalian cells: their biophysical, molecular, and pharmacological properties. Pharmacol Rev 2019; 71: 49-88.
48. Song Y, Wang L, Wang J, et al. Aquaporins in Respiratory System. In: Yang B. (eds), Aquaporins. Advances in Experimental Medicine and Biology. Dordrecht: Springer, 2017, vol. 969, p 115-22.
49. Zhang ZQ, Song YL, Chen ZH, et al. Deletion of aquaporin 5 aggravates acute lung injury induced by Pseudomonas aeruginosa. J Trauma 2011; 71: 1305-11.
50. Martin SL, Saint-Criq V, Hwang TC, et al. Ion channels as targets to treat cystic fibrosis lung disease. J Cyst Fibros 2018; 17(2S): S22-S27
51. Saint-Criq V, Gray MA. Role of CFTR in epithelial physiology. Cell Mol Life Sci 2017; 74: 93-115.
52. Fajac I,Wainwright CE. New treatments targeting the basic defects in cystic fibrosis. Presse Med 2017; 46: e165-75.
53. Fajac I, De Boeckc K. New horizons for cystic fibrosis treatment. Pharmacol Ther 2017; 170: 205-11.
54. Solomon GM, Fu L, Rowe SM, et al. The therapeutic potential of CFTR modulators for COPD and other airway diseases. Curr Opin Pharmacol 2017, 34: 132-9.
55. Perry LA, Penny-Dimri JC, Aslam AA, et al. Topical cystic 314 MEDICINA – Volumen 79 – Nº 4, 2019 fibrosis transmembrane conductance regulator gene replacement for cystic fibrosis-related lung disease. Paediatr Respir Rev 2017; 22: 47-4.
56. Solomon GM, Fu LW, Rowe STM, et al. The therapeutic potential of CFTR modulators for COPD and other airway diseases. Curr Opin Pharmacol 2017; 34: 132-9.
57. Solomon GM, Hathorne H, Liu B, et al. Pilot evaluation of the CFTR potentiator ivacaftor for the treatment of chronic bronchitis. Lancet Respir Med 2016; 4: e32-3.
58. Ding Y, Zhao R, Zhao X, Matthay MA, Nie H-G, Ji H-L. ENaCs as both effectors and regulators of MiRNAs in lung epithelial development and regeneration. Cell Physiol Biochem 2017; 44: 1120-32.
59. Nakao L, Kanaji S, Otha S, et al. Identification of pendrin as a common mediator for mucus production in bronchial asthma and chronic obstructive pulmonary disease. J Immunol 2008; 180: 6262-9.
60. Callaghan RM, Voynow JA. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev 2006; 86: 245-78.
61. Fernández Fernández E, De Santi C, De Rose V, et al. CFTR dysfunction in cystic fibrosis and chronic obstructive pulmonary disease. Expert Rev Respir Med 2018; 12: 483-92.
62. Brett TJ. CLCA1 and TMEM16A: the link towards a potential cure for airway diseases. Expert Rev Respir Med 2015; 9: 503-6.
63. Romero MF, Chen AP, Parker MD, et al. The SLC4 family of bicarbonate (HCO3-) transporters. Mol Aspects Med 2013; 34: 159-82.
64. Arroyo JP, Kahle KT, Gamba G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol Aspects Med 2013; 34: 288-98.
65. Alper SL, Sahrma AK. The SLC26 gene family of anion transporters and channels. Mol Aspects Med 2013; 34: 494-515.
– – – –
Michel de Montaigne (1533-1592)
Ensayos (1580-1588). De la fuerza de la imaginación. Libro I XX, III, p61. Traducción de Juan G. de Luaces. Buenos Aires; Hyspamérica, 1984