
RENÉE CRISP 1 *, YESICA BESTACH 2, LAURA KORNBLIHTT 3 *, CAROLINA LAZZARINO 4 *, MARCELO IASTREBNER 5 *, JAQUELINE GONZALEZ 6 *, JORGE ARBELBIDE 7 *, CAROLINA B. BELLI 2 *
1 Servicio de Hematología, Hospital Nacional Prof. Alejandro Posadas, Haedo, 2Laboratorio de Genética Hematológica, Instituto de Medicina Experimental (IMEX-CONICET)/Academia Nacional de Medicina, 3 División Hematología, Hospital de Clínicas José de San Martín, Universidad de Buenos Aires, 4 Servicio de Hematología, Hospital Zonal de Agudos Dr. Diego Paroissien, Isidro Casanova, 5 Servicio de Hematología, Sanatorio Sagrado Corazón, 6 Servicio de Hematología, Hospital General de Agudos Carlos G. Durand, 7 Sección Hematología, Hospital Italiano, Buenos Aires, Argentina
*En representación del Grupo de Estudio de Síndromes Mielodisplásicos, Sociedad Argentina de Hematología
Resumen La Argentina es un país caracterizado por una distribución heterogénea de su población, de sus recursos económicos y, consiguientemente, del acceso a los servicios de salud, lo cual podría afectar el diagnóstico y tratamiento de los pacientes con síndromes mielodisplásicos. Basados en la complejidad creciente para arribar al diagnóstico, estimar el riesgo e indicar un tratamiento adecuado, hemos conducido una encuesta de veintitrés preguntas para evaluar patrones de práctica clínica. El cuestionario se distribuyó entre los 850 hematólogos argentinos inscriptos al XXII Congreso Argentino de Hematología y 195 (22.9%) fueron contestados. El 40.0% refieren que < 75% de sus pacientes acceden al cariotipo, histología de la médula ósea y citometría de flujo. Este acceso disminuye significativamente por una baja cobertura sanitaria (OR 6.3), en población adulta (OR 3.8), al derivar el estudio citogenético (OR 3.2) y fuera del área metropolitana de Buenos Aires (OR 2.4). Los encuestados evitan terminologías oncológicas (77.0%) al introducir el diagnóstico y utilizan el sistema internacional de predicción o su revisión (74.2%) para estadificar riesgo. Sin embargo, éstos priorizan la edad al seleccionar tratamiento y los pediatras indican preferentemente el trasplante de precursores hematopoyéticos. La mayoría de los hematólogos ha prescripto los tratamientos recomendados, cuyas suspensiones se relacionaron con falta de respuesta (62.7%), con participación reducida en ensayos clínicos (8.9%). Por ende, refieren heterogeneidad en el acceso a las herramientas diagnósticas complementarias con diferencias al momento de indicar un tratamiento, dependiendo de la edad de sus pacientes, sin limitaciones aparentes en su prescripción.
Palabras clave: síndromes mielodisplásicos, histología de médula ósea, encuesta de salud, patrones de práctica clínica
Abstract Preferences and limitations of hematologists to address the complexity of myelodysplastic syndromes. Argentina is a country characterized by a heterogeneous distribution of its population, its economic resources and, consequently, access to health services, which could affect the diagnosis and treatment of patients with myelodysplastic syndromes. Based on the increasing complexity to arrive at the diagnosis, estimate the risk and indicate an adequate treatment, we have conducted a survey of twenty-three questions to evaluate patterns of clinical practice. The questionnaire was distributed among 850 hematologists registered at the XXII Argentine Congress of Hematology, and 195 (22.9%) were answered; 40.0% report that < 75% of their patients access the karyotype, bone marrow histology and flow cytometry. This access decreases significantly due to low health coverage (OR 6.3), in the adult population (OR 3.8), when the cytogenetic study is derived (OR 3.2) and outside the metropolitan area of Buenos Aires (OR 2.4). The respondents avoid oncological terminologies (77.0%) when introducing the diagnosis and use the international prediction system or its review (74.2%) to stage risk. However, they prioritize age when selecting treatment and pediatricians preferentially recommend the transplantation of hematopoietic precursors. Most of the haematologists have prescribed the recommended treatments, whose suspensions were related to lack of response (62.7%), with reduced participation in clinical trials (8.9%). Therefore, they report heterogeneity in the access to complementary diagnostic tools with differences at the time of indicating a treatment, depending on the age of their patients without apparent limitations in their prescription.
Key words: myelodysplastic syndromes, bone marrow histology, physician practice patterns, health survey
Dirección postal: Carolina Belli, Laboratorio de Genética Hematológica, IMEX-CONICET/Academia Nacional de Medicina, Pacheco de Melo 3081, 1425 Buenos Aires, Argentina
e-mail: cbelli@hematologia.anm.edu.ar
Los síndromes mielodisplásicos (SMD) son un grupo heterogéneo de enfermedades clonales caracterizadas por la presencia de citopenia(s), hematopoyesis ineficaz y riesgo de progresión a leucemia mieloide aguda (LMA) 1.
La incidencia varía de 3-5/100 000 individuos por año, con un incremento asociado a la edad de 0.5-4.0/1 000 000 en niños a 20/100 000 en mayores de 70 años 2-4. Otras publicaciones han duplicado estos hallazgos 5 alcanzando un estimado de 103/100 000 en mayores de 64 años 6. Estos últimos cálculos posicionarían a los SMD entre las diez neoplasias más frecuentes en población adulta mayor 6. La subestimación reflejada en los registros podría relacionarse a la omisión de casos sintomáticos sin confirmación diagnóstica, a demoras en la derivación a un médico hematólogo o a la denuncia de casos bajo denominaciones no reconocidas 6-8. Los algoritmos de diagnóstico y clasificación propuestos por la Organización Mundial de la Salud (OMS) en 2016 requieren de un escenario multidisciplinario: profesionales experimentados en el reconocimiento de los hallazgos displásicos y el recuento de blastos, citogenetistas y biólogos moleculares, al incluirse la presencia de mutaciones en el gen Splicing Factor 3b Subunit 1 (SF3B1)1. El examen histológico de la médula ósea (EHMO) y la determinación del inmunofenotipo mediante citometría de flujo multiparamétrica (IF-CFM) pueden colaborar en el diagnóstico 9-11.
El comportamiento clínico también es heterogéneo: un tercio de los pacientes alcanzan una sobrevida comparable a la esperada para su edad; mientras que, otros fallecen a los pocos meses del diagnóstico por causas asociadas a su falla medular, con o sin evolución a LMA 1, 12. Por lo tanto, la determinación del pronóstico y la selección del tratamiento requieren de una cuidadosa estratificación del riesgo 1, 12. Los abordajes terapéuticos varían desde cuidados de soporte hasta el trasplante de células precursoras hematopoyéticas (TCPH) 12. Las variables tomadas en consideración incluyen aquellas relacionadas a la gravedad de la enfermedad: profundidad de la(s) citopenia(s), porcentaje de blastos y cariotipo, las cuales componen el esqueleto de los sistemas de predicción, como el Sistema de Puntuación de Pronóstico Internacional (IPSS, por sus siglas del inglés)13 o su versión revisada, el IPSS-R 14. Sin embargo, otros factores propios del caso, como la dependencia transfusional, la edad y la presencia de comorbilidades, también son tomados en consideración 15.
La Argentina es un país caracterizado por una distribución heterogénea de su población, de sus recursos económicos y, consiguientemente, del acceso a los servicios de salud 16, lo cual podría afectar el diagnóstico y tratamiento de los pacientes con SMD. Basados en la complejidad creciente para arribar al diagnóstico, estimar el riesgo e indicar un tratamiento acorde hemos conducido una encuesta a fin de evaluar la distribución de las herramientas diagnóstico-terapéuticas con las que cuenta el médico hematólogo.
Materiales y métodos
La encuesta se realizó mediante un cuestionario impreso diseñado por la Subcomisión de SMD de la Sociedad Argentina de Hematología (SAH) y optimizado en base a una prueba piloto en la que participaron 28 médicos hematólogos.
Las 23 preguntas cerradas de opción múltiple incluyeron diversos aspectos concernientes a la práctica profesional en general y, en referencia a los pacientes con SMD, se evaluó el acceso a los métodos diagnósticos, la forma en que la enfermedad se presenta a los pacientes, la elección de sistemas pronósticos y las estrategias terapéuticas (Anexo).
El cuestionario fue distribuido entre los profesionales médicos presentes en el XXII Congreso Argentino de Hematología, 2015, y la participación fue voluntaria y anónima. Las respuestas válidas fueron analizadas de manera descriptiva y, en caso de comparaciones se aplicaron las pruebas de Chi2, el test exacto de Fisher y análisis de regresión logística utilizando el método de entrada. Los resultados fueron expresados en porcentajes relativos, como razón de razones (Odds Ratio: OR) con el respectivo intervalo de confianza (IC) 95% y se fijó el nivel de significación en 0.05. Para el análisis se utilizó el programa SPSS v.17 (SPSS, Chicago, EE.UU.) y el GraphPad Prism versión 5.00 (GraphPad Software, San Diego, EE.UU.).
Resultados
La tasa de respuesta fue del 22.9%: 195 encuestas obtenidas de 850 hematólogos argentinos inscriptos. El 55.7% de los encuestados ha atendido pacientes con SMD por más de 10 años con un predominio (78.5%) especializado en población adulta. Dos tercios (66.7%) ejercía en el ámbito público, el 38.7% combinaba sus actividades y un 3.6% atendía únicamente en consultorios particulares. El 34.9% de los hematólogos refirieron que > 75% de sus pacientes poseían cobertura en salud. La mayoría atendían menos de 10 pacientes (84.6%) de bajo riesgo y menos de 5 (79.6%) de alto riesgo (Tabla 1).
A fin de establecer el diagnóstico el 76.5% solicita el estudio citogenético, la examinación histológica y el IFCFM de la MO (Tabla 2). Al analizar las 22 respuestas ambiguas sí/no para el requerimiento del estudio citogenético, la mayoría poseía una experiencia = 10 años (68.2%), ejercían en el ámbito público (86.7%) combinando con otras modalidades de atención (42.1%), atendían población adulta (77.3%) con una cobertura asistencial < 51% (68.2%), similar a lo observado para las prácticas restantes.
La examinación histológica resultó como la práctica más accesible y realizada con la mayor frecuencia en el mismo lugar de atención (p < 0.001) por patólogos con o sin especialidad. Los hematopatólogos fueron mayoritariamente seleccionados por los médicos de adultos (p < 0.001, OR 7.39), en caso de derivación (p = 0.009, OR 3.95) y pertenecientes al área metropolitana (p<0.001, OR 3.95).
Los factores que influyeron independientemente en una accesibilidad menor al 75% a los tres estudios complementarios (40.0% de las respuestas) fueron: baja cobertura médico-asistencial (22.1% vs. 49.6%, OR 6.34), población adulta (17.9% vs. 42.5%, OR 3.84), derivación del estudio citogenético (34.2% vs. 47.4%, OR 2.38) y ejercer profesionalmente fuera del área metropolitana (34.2% vs. 47.4%, OR 2.38) (Fig. 1).
El panel completo de estudios moleculares no se realizaba en el país al momento de la encuesta y solo el 18.0% respondió tener acceso a ciertas determinaciones. Aunque la inclusión de los hallazgos moleculares en los sistemas de predicción de riesgo fue considerada de interés clínico por el 58.2%, el 50.8% percibía que sus pacientes no tendrán acceso en los próximos cinco años y el 40.8% que es solo para investigación o accesible en centros importantes norteamericanos o europeos.
Los hematólogos seleccionaron que introducen el diagnóstico de SMD como una falla medular (43.1%) y, en menor medida, como estado preleucémico (17.4%), enfermedad de la sangre (12.8%) y el 5.6% como cáncer/neoplasia. La elección hacia la falla medular se incremen-ta al 47.2% y las “neoplásicas” al 31% cuando se computan las selecciones múltiples (17.2%). La terminología empleada fue independiente del perfil profesional.

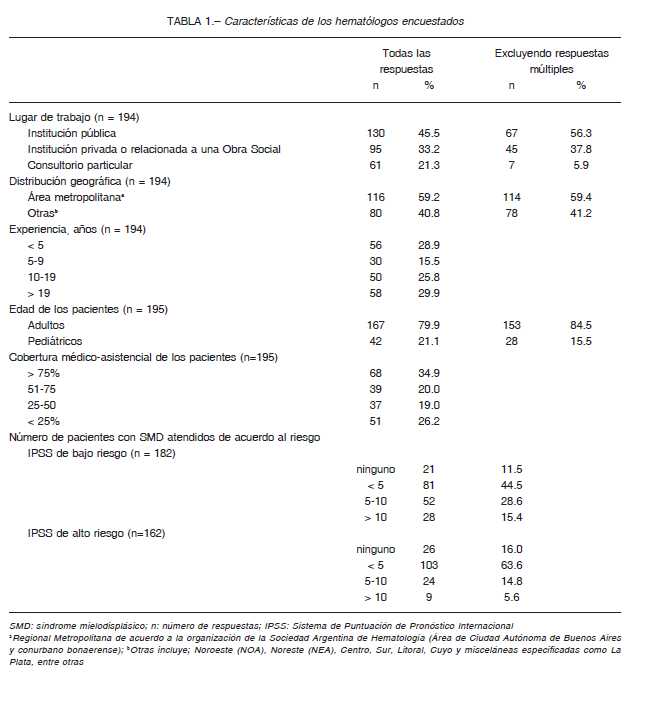
Los sistemas IPSS (33.3%) e IPSS-R (30.6%) fueron preferidos. Sin embargo, el 31.1% aplican múltiples sistemas de predicción y la mayoría priorizan la edad de los pacientes al decidir la estrategia terapéutica. Los demás factores considerados dependieron de la edad (p = 0.004): el IPSS y la presencia de comorbilidades resaltan entre los hematólogos de adultos y el perfil fue más homogéneo entre los pediatras (Fig. 2).
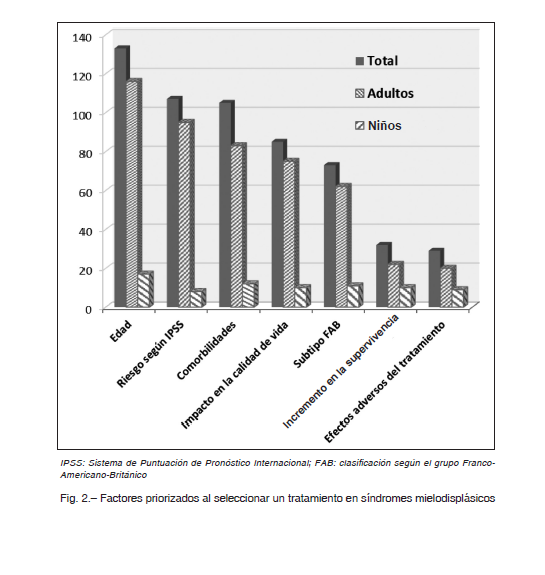
La mayoría de los profesionales informan a sus pacientes todas las estrategias terapéuticas (59.9%) o, al menos, las que consideran necesario administrar (36.5%), y solo un 3.1% restringe la información a aquellas que tiene la oportunidad de indicar. En la Figura 3 se observa que más del 90% ha indicado transfusiones de glóbulos rojos (TGR) y de plaquetas, eritropoyetina (EPO) y 5-azacitidina (AZA). La terapia quelante de hierro (73.1%), 5-aza-2’-deoxicitidina (DAC) (59.0%), lenalidomida (LEN) (54.0%), trasplante de células progenitoras hematopoyéticas (TCPH) (50.0%) y ensayos clínicos (8.9%) fueron seleccionados en menor medida.
Las indicaciones de tratamiento variaron de acuerdo a la edad de los pacientes: EPO ± factores de crecimiento, AZA, DAC y LEN fueron seleccionados con menor frecuencia por los pediatras (p = 0.002, p = 0.048, p = 0.067, p = 0.007 y p = 0.022, respectivamente) en quienes resalta el TCPH (90.9% vs. 43.9%, p < 0.001).
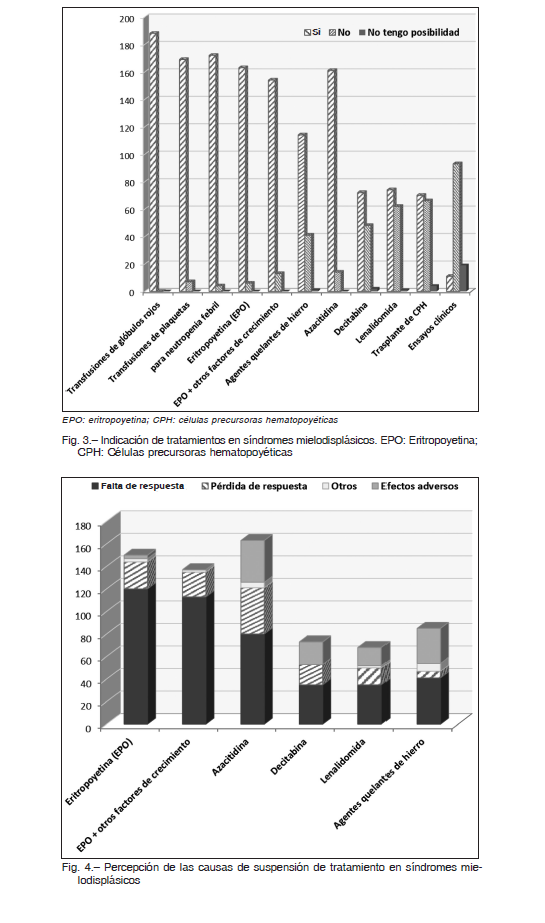
La mayoría de los hematólogos respondieron que administran de 4 a 6 ciclos de AZA (68.3%), DAC (77.3%) o LEN (56.0%), o más ciclos (25.2%, 14.3% y 14.3%, respectivamente), antes de decidir que no se alcanza una respuesta adecuada. Según el criterio general de los encuestados, la suspensión del tratamiento se relaciona a una falta de respuesta (62.7%) la cual fue mayoritariamente elegida frente a la utilización de EPO (80.0%) ± factores de crecimiento (82.5%). La presencia de efectos adversos y la pérdida de la respuesta fueron similares en relación a la utilización de AZA, DAC o LEN, mientras que, la presencia de efectos adversos fue mayor frente a la terapia quelante (36.5%) (Fig. 4).
Discusión
Ciertas características clínicas y diversos factores de riesgo, incluyendo la validación de sistemas de predicción y el resultado del TCPH, han sido previamente descriptos en diversas series locales de pacientes con SMD 17-20. El presente estudio explora los patrones de práctica clínica en una muestra representativa de hematólogos activamente involucrados en el diagnóstico y tratamiento de estas enfermedades, tanto en adultos como en niños.
La evaluación morfológica de la sangre periférica y de la MO en pacientes con sospecha de SMD se encuentra dentro de las incumbencias de los médicos hematólogos en la Argentina y ellos indican estudios complementarios conforme a las recomendaciones vigentes 1, 9, 12, 21. En otros países esta práctica es realizada por patólogos 22, quienes en nuestro medio, examinan la biopsia de MO colaborando con información relevante para el diagnóstico y pronóstico 9-10. Este estudio es muy solicitado por los encuestados, similar a lo relevado por el Instituto Nacional del Cáncer de EE.UU. 23. La alta accesibilidad estaría directamente relacionada con su disponibilidad en el centro de atención, realizada por especialistas en la mitad de los casos. Según patólogos encuestados previamente, solo un tercio confiaba en su habilidad para distinguir un SMD de otros desórdenes hematológicos, y sus falencias abarcaban aspectos de formación y práctica, las cuales se reducían entre los hematopatólogos 22. De acuerdo a los datos relevados, estos especialistas se encuentran mayoritariamente centralizados y son preferidos por los hematólogos de adultos en sus derivaciones.
La información acerca de las preferencias en cuanto al IF-CFM es limitada. Según el estudio anteriormente mencionado, el análisis de los blastos y de granulocitos sería solicitado solo por un sexto de los patólogos 22. Este porcentaje fue ampliamente superado en la presente encuesta y la mayoría de los pacientes accedería, aunque requieran derivación. Esta técnica ha ido ganando adeptos y algunos grupos proponen integrarla en los algoritmos de la OMS 11. El cariotipo es muy demandado ya que es esencial para el establecimiento del diagnóstico y la selección de terapias adaptadas a riesgo 13-15, 21. El porcentaje de pacientes estudiados varía en las diferentes series entre 50%-90% 13, 14, 17-19, 23-25 y disminuye significativamente en los mayores de 75 años en el registro germano 23. La encuesta describe que su disponibilidad es significativamente inferior al de la biopsia y que es mayoritariamente derivado, dificultando la interpretación multidisciplinaria e integral de los resultados. En un análisis previo se describió una fuerte centralización de hematólogos, reflejando la distribución de las regionales en la SAH, con un acceso heterogéneo de las herramientas necesarias 26. En el presente estudio, gracias al abordaje multivariado, pudo confirmarse que ejercer por fuera del área metropolitana limita el acceso a las tres herramientas complementarias. Además, esta disponibilidad se vio negativamente afectada por una baja cobertura médico-asistencial, la necesidad de derivar el estudio citogenético y atender pacientes adultos. Por ende, la conjunción de estos factores independientes disminuye el acceso al diagnóstico adecuado y a la estratificación de riesgo apropiada. Los hematólogos prefieren, al igual que en estudios previos 7, 27-29, evitar las palabras cáncer/ neoplasia o leucemia cuando introducen el concepto de SMD a sus pacientes. Consecuentemente, éstos y sus cuidadores han referido que la información recibida es insuficiente o inadecuada 29, que entienden poco de la enfermedad 27-29 y un tercio desconoce su riesgo de progresión leucémica 30.
Esta introducción optimista puede impactar en la adherencia a las consultas, a estudios rutinarios y al tratamiento, si lo hubiera. Una definición no neoplásica se traduce en consecuencias prácticas como falta de reclamo por cobertura y en la calidad del dato de los registros 7. Este alto porcentaje podría reflejar que la mayoría de los casos que atienden los encuestados son de bajo riesgo, conforme a las series publicadas por nuestro grupo 17-19. Sin embargo, el bajo riesgo requiere monitoreo, con una sobrevida media de 2-6 años y causas de muerte mayoritariamente asociadas 13-14, 17-19. Por otro lado, las terminologías “neoplásicas” se incrementan al incluir las respuestas múltiples, lo cual podría interpretarse en elecciones alternativas de terminologías dependientes de las características de los pacientes.
La heterogeneidad clínica de la enfermedad requiere de una cuidadosa estratificación de riesgo a fin de aplicar los algoritmos terapéuticos 12, 21. La mayoría de los encuestados eligen el IPSS 13 o el IPSS-R 14, basados en parámetros relacionados a la enfermedad. Sin embargo, priorizan características de los enfermos, como la edad o las comorbilidades, al indicar un tratamiento, lo cual explicaría el uso combinado de diversos sistemas. Los hallazgos moleculares colaboran en la caracterización biológica y, aunque no hay consenso, en la predicción de riesgo y respuesta al tratamiento 31-32. El panel completo por técnicas de nueva generación no estaba disponible en nuestro medio al momento de la encuesta y la mayoría de los encuestados consideraron que no podrán acceder en los próximos cinco años o que son de acceso restringido al primer mundo, limitando la aplicabilidad de sistemas de predicción basados en hallazgos moleculares.
La mayoría de los encuestados introducen todas las alternativas terapéuticas y un tercio las que consideran administrar. Esta última opción ha sido descrita como la preferida por pacientes y sus cuidadores a fin de evitar la sobrecarga de información 33. El requerimiento de transfusiones de plaquetas o glóbulos rojos (GR) se incrementa del 6 al 20% en los de bajo riesgo hasta el 33-67% de los de riesgo alto 25, con una incidencia acumulada a los 3 años del 45% en pacientes mayores de 66 años 34. Ambas opciones son ampliamente indicadas por los hematólogos a fin de enfrentar la(s) citopenia(s) sintomática(s). Los enfermos dependientes de transfusiones de GR poseen un riesgo incrementado de sobrecarga de hierro cuyo tratamiento con agentes quelantes ha sido indicado por tres cuartos de los participantes, asociado con la mayor tasa de interrupción por efectos adversos, coincidente con una encuesta previa 35. La eritropoyetina (EPO) ± factores de crecimiento también fue considerablemente seleccionada, como fuera anteriormente descripto 25, 27, en gran parte para población adulta y asociada a la menor tasa de respuesta como causa de suspensión.
La lenalidomida, disponible desde 2008 y recomendada para casos de bajo riesgo con la alteración citogenética del (5q) 12, 21, ha sido indicada por la mitad de los encuestados cuya mayor causa de interrupción fue la falta de respuesta. Entre los agentes hipometilantes, los hematólogos han prescripto más frecuentemente AZA, disponible desde 2007, que decitabina (DAC), desde 2008, y ambos también asociados a falta de respuesta como mayor causa de suspensión. Estos agentes fueron elegidos previamente como primera opción para todo paciente de alto riesgo, seguidos por el TCPH para pacientes candidatos 36. Su uso también se recomienda en casos de bajo riesgo, dependientes de transfusiones y resistentes a EPO 12, 21.
El TCPH es la única opción con potencial curativo y fue preferentemente elegida por los pediatras. Su menor indicación por los encuestados especializados en adultos puede estar asociada a la edad de los casos, con una mediana de 70 años 17-19, y a la importancia que adjudicaron a las comorbilidades al seleccionar un tratamiento. Aunque el 44% refiere haber indicado un trasplante, en nuestra experiencia, solo el 6% de los pacientes accedieron18 con un límite de edad de 65 años 20. Esta baja tasa concuerda con datos de encuestas previas donde solo el 4% de los enfermos lo reconocieron como opción o habían sido evaluados para el procedimiento 7, 25, 28. Además, las complicaciones asociadas pueden influir en el bajo interés por este tratamiento 28, lo que sugeriría que el mismo pueda no ser ofrecido a pacientes candidatos 7.
En la última década se ha avanzado en el conocimiento de la biología de la enfermedad, pero sin nuevas opciones terapéuticas. De acuerdo a nuestros resultados es posible especular que los encuestados evitan la palabra cáncer/leucemia a fin de disminuir los niveles de ansiedad de sus enfermos, quienes poseen poca o ninguna opción curativa y son, frecuentemente, adultos mayores y de bajo riesgo 17-19. Pacientes que conocen su riesgo de evolución participarían de ensayos clínicos hipotéticos 30. Sin embargo, una minoría de los encuestados ha ofrecido estos ensayos, a diferencia de otros contextos 6.
Por ende, los hematólogos refieren dificultades en el acceso a las herramientas diagnósticas complementarias y diferencias al momento de seleccionar una estrategia terapéutica dependiendo de la edad, sin limitaciones aparentes para prescribir los tratamientos recomendados y con acceso limitado en la participación de ensayos clínicos.
Agradecimientos: A los miembros del Grupo de Estudio de SMD de la SAH por su participación en la distribución del cuestionario y en la discusión de los resultados.
Este trabajo no fue específicamente subsidiado. Sin embargo, los autores reciben subsidios de la Agencia Nacional de Promoción Científica y Tecnológica [C.B. y M.G.F., PID 0044 y C.B., PICT 0480] y del Consejo Nacional de Investigaciones Científicas y Técnicas [C.B., PIP 0056].
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127: 2391-405.
2. Passmore SJ, Chessells JM, Kempski H, et al. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol 2003; 121: 758-67.
3. Aul C, Gattermann N, Schneider W. Age-related incidence and other epidemiological aspects of myelodysplastic syndromes. Br J Haematol 1992; 82: 358-67.
4. Williamson PJ, Kruger AR, Reynolds PJ, Hamblin TJ, Oscier DG. Establishing the incidence of myelodysplastic syndrome. Br J Haematol 1994; 87: 743-5.
5. De Roos AJ, Deeg HJ, Onstad L, et al. Incidence of myelodysplastic syndromes within a nonprofit healthcare system in western Washington state, 2005-2006. Am J Hematol 2010; 85: 765-70.
6. McQuilten ZK, Wood EM, Polizzotto MN, et al. Underestimation of myelodysplastic syndrome incidence by cancer registries: Results from a population-based data linkage study. Cancer 2014; 120: 1686-94.
7. Steensma DP, Komrokji RS, Stone RM, et al. Disparity in perceptions of disease characteristics, treatment effectiveness, and factors influencing treatment adherence between physicians and patients with myelodysplastic syndromes. Cancer 2014; 120: 1670-6.
8. Shammo JM, Foran JM, Houk A, et al. An examination of educational gaps in the diagnosis and treatment of myelodysplastic syndromes. Cancer Control 2011; 18: 65-74.
9. Valent P, Horny HP, Bennett JM, et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: Consensus statements and report from a working conference. Leuk Res 2007; 31: 727-36.
10. Della Porta MG, Malcovati L, Boveri E, et al. Clinical relevance of bone marrow fibrosis and CD34-positive cell clusters in primary myelodysplastic syndromes. J Clin Oncol 2009; 27: 754-62.
11. Porwit A, van de Loosdrecht AA, Bettelheim P, et al. Revisiting guidelines for integration of flow cytometry results in the WHO classification of myelodysplastic syndromesproposal from the International/European Leukemia Net Working Group for Flow Cytometry in MDS. Leukemia 2014; 28: 1793-8.
12. Garcia-Manero G. Myelodysplastic syndromes: 2015 Update on diagnosis, risk-stratification and management. Am J Hematol 2015; 90: 831-41.
13. Greenberg P, Cox C, LeBeau M, et al. International Scor ing System for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89: 2079-88.
14. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012; 120: 2454-65.
15. Germing U, Kündgen A. Prognostic scoring systems in MDS. Leuk Res 2012; 36: 1463-9.
16. INDEC. Instituto Nacional de Estadísticas y Censos. Censo Nacional de Población, Hogares y Viviendas 2010. Censo Del Bicentenario Resultados Definitivos. Serie B Nº 2, Buenos Aires: Talleres Gráficos Prix, 2012.
17. Belli CB, Bestach Y, Giunta M, et al. Application of the revised International Prognostic Scoring System for myelodysplastic syndromes in Argentinean patients. Ann Hematol 2014; 93: 705-7.
18. Belli CB, Pinheiro RF, Bestach Y, et al. Myelodysplastic syndromes in South America: a multinational study of 1080 patients. Am J Hematol 2015; 90: 851-8.
19. Enrico A, Bestach Y, Flores MG, et al. Influence of acute myeloid leukemia progression on the prognosis of 831 patients with myelodysplastic syndromes from the Argentine database. Clin Lymphoma Myeloma Leuk 2017;17: 743-2.
20. Basquiera AL, Rivas MM, Remaggi G, et al. Allogeneic hematopoietic stem cell transplantation in adults with myelodysplastic syndrome: Experience of the Argentinean Group of Bone Marrow Transplantation (GATMO). Hematology 2016; 21: 162-9.
21. Alfonso G, Arbelbide J, Basquiera AL, et al. Síndromes mielodisplásicos y síndromes de superposición neoplasias mieloproliferativas/ síndromes mielodisplásicos. En: Sociedad Argentina de Hematología, eds. Guía de Diagnóstico y Tratamiento de la Sociedad Argentina de Hematología, 2017 ed. Buenos Aires: Estudio Sigma SRL, 2017, p 611-38.
22. Glauser TA, Sagatys EM, Williamson JC, et al. Current pathology practices in and barriers to MDS diagnosis. Leuk Res 2013; 37: 1656-61.
23. Gattermann N, Kündgen A, Kellermann L, et al. The impact of age on the diagnosis and therapy of myelodysplastic syndromes: results from a retrospective multicenter analysis in Germany. Eur J Haematol 2013; 91: 473-82.
24. Craig BM, Rollison DE, List AF, Cogle CR. Diagnostic testing, treatment, cost of care, and survival among registered and non-registered patients with myelodysplastic syndromes. Leuk Res 2011; 35: 1453-6.
25. Sekeres MA, Schoonen WM, Kantarjian H, et al. Characteristics of US patients with myelodysplastic syndromes: results of six cross-sectional physician surveys. J Natl Cancer Inst 2008; 100: 1542-51.
26. Crisp R, Flores MG, Enrico A, et al. Accesibilidad de herramientas diagnóstico-terapéuticas para los pacientes con síndromes mielodisplásicos en la República Argentina. Hematología 2017; 21: 127-38.
27. Sekeres MA, Maciejewski JP, List AF, et al. Perceptions of disease state, treatment outcomes, and prognosis among patients with myelodysplastic syndromes: results from an Internet-based survey. Oncologist 2011; 16: 904-11.
28. Smith BD. Myelodysplastic syndromes: challenges to improving patient and caregiver satisfaction. Am J Med 2012; 125: S26-30.
29. Besson C, Rannou S, Elmaaroufi H, et al. Disclosure of myelodysplastic syndrome diagnosis: improving patients’ understanding and experience. Eur J Haematol 2013; 90: 151-6.
30. Ousseine YM, Butow PN, Julian-Reynier C, et al. Awareness of acute myeloid leukaemia risk induced by diagnosis of a myelodysplastic syndrome. Leuk Res 2016; 46: 79-84.
31. Reinig E, Yang F, Traer E, et al. Targeted next-generation sequencing in myelodysplastic syndrome and chronic myelomonocytic leukemia aids diagnosis in challenging cases and identifies frequent spliceosome mutations in transformed acute myeloid leukemia. Am J Clin Pathol 2016; 145: 497-506.
32. Visconte V, Tabarroki A, Gerace CJ, et al. Screening for SF3B1 mutations is a useful tool to differentiate between acquired clonal and non-clonal sideroblastic anemia. Leuk Lymphoma 2015; 56: 1888-90.
33. Mancini J, Butow PN, Julian-Reynier C, et al. Question prompt list responds to information needs of myelodysplastic syndromes patients and caregivers. Leuk Res 2015; 39: 599-605.
34. Lindquist KJ, Danese MD, et al. Health care utilization and mortality among elderly patients with myelodysplastic syndromes. Ann Oncol 2011; 22: 1181-8.
35. Giagounidis A, Leto di Priolo S, et al. A European survey on the detection and management of iron overload in transfusion-dependent patients with MDS. Ann Hematol 2011; 90: 667-73.
36. Sohn SK, Moon JH, Lee YJ, et al. Survey of experts on therapeutic policies and proposals for the optimal timing for allogeneic peripheral blood stem cell transplantation in transfusion-dependent patients with myelodysplastic syndrome-refractory anemia. Blood Res 2016; 51: 44-9.

