
TOMÁS DALOTTO-MORENO1*, ADA G. BLIDNER1*, M. ROMINA GIROTTI1, 2,*,
SEBASTIÁN M. MALLER1, GABRIEL A. RABINOVICH1-3
1Laboratorio de Inmunopatología, Instituto de Biología y Medicina Experimental (IBYME), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), 2Laboratorio de Inmuno-Oncología Translacional, Instituto de Biología y Medicina Experimental (IBYME), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Buenos Aires, Argentina, 3Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina
*Contribuyeron de igual forma a la elaboración de este trabajo
Resumen La activación del sistema inmunológico en pacientes con cáncer ha sido un objetivo histórico en el campo de la oncología. En las últimas décadas, nuestro entendimiento de la respuesta inmunológica antitumoral ha promovido el desarrollo de novedosas estrategias terapéuticas dando como resultado un cambio de paradigma en el tratamiento del cáncer. La utilización de agentes bloqueantes de puntos de chequeo del sistema inmunológico como PD-1/PD-L1 y CTLA-4, de agonistas de moléculas co-estimuladoras como CD137 y OX-40 y la transferencia adoptiva de células T antitumorales modificadas genéticamente han generado importantes beneficios clínicos, reflejados en respuestas objetivas y durader as, en enfermos sin tratamientos convencionales disponibles. Sin embargo, un gran número de pacientes no responde a dichas terapias generando resistencia o sufriendo recaídas de la enfermedad debido a la aparición de circuitos inhibitorios o compensatorios. La combinación racional de estrategias terapéuticas permite eliminar mecanismos de resistencia, mientras que la identificación de biomarcadores predictivos facilita la selección de pacientes respondedores a dichos tratamientos. Recientes ensayos clínicos y estudios pre-clínicos permiten vislumbrar un escenario optimista con importantes desafíos en la implementación de estrategias de inmunoterapia en cáncer.
Palabras clave: inmunoterapia, cáncer, puntos de chequeo inmunológico, receptores quiméricos de antígeno, co-estimulación, biomarcadores
Abstract Immunotherapy in cancer. Current prospects, challenges and new horizons. Recent under-standing of the mechanisms that control immune system homeostasis and orchestrate antitumor responses has prompted the development of novel immunotherapeutic modalities. These include antibodies that target immune checkpoints such as PD-1/PD-L1 and CTLA-4, agonistic antibodies of costimulatory molecules such as CD137 and OX-40 and the adoptive transfer of genetically-modified antitumor T cells. However, a large number of patients do not respond to these therapies and develop resistance as a result of activation of compensatory circuits. Rational combination of immunotherapeutic modalities will help overcome resistance and will increase the number of patients who will benefit from these treatments. Moreover, identification of predictive biomarkers will allow selection of patients responding to these treatments. Emerging clinical trials and pre-clinical studies have shown exciting results anticipating new horizons in the design and implementation of cancer immunotherapeutic modalities.
Key words: immunotherapy, cancer, immunological checkpoints, chimeric antigen receptors, co-stimulation, biomarkers
Recibido: 23-VII-2018 Aceptado: 22-VIII-2018
Dirección postal: Gabriel A. Rabinovich, Laboratorio de Inmunopatología, Instituto de Biología y Medicina Experimental (IBYME), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Vuelta de Obligado 2490, 1428 Buenos Aires, Argentina
e-mail: gabriel.r@ibyme.conicet.gov.ar
La estimulación del sistema inmunológico para el tratamiento contra el cáncer tiene sus orígenes hacia finales del siglo XIX, gracias a los experimentos del cirujano y oncólogo William Coley (Nueva York), quien utilizaba toxinas derivadas de bacterias Streptococcus erysipelas y Bacillus prodigiosus para el tratamiento de un tipo de sarcoma inoperable. Si bien los mecanismos subyacentes a este efecto no se conocían en ese momento, fue posible postular que la inflamación local producida por estas toxinas era capaz de reactivar la respuesta inmunológica y eliminar el tumor. Estos primeros estudios sentaron las bases de lo que en la actualidad constituye una revolución en el tratamiento del cáncer1.
Terapias basadas en el bloqueo de puntos de control (checkpoints) inmunológicos
La historia de la inmunoterapia del cáncer prosigue a fines de 1980 con el tratamiento local y sistémico con citoquinas (IL-2, IFN-g, TNF); sin embargo, fue a principios de 2011 que la inmunoterapia cobró relevancia a nivel mundial con el advenimiento del bloqueo de los puntos de control (checkpoints) inmunológicos. La lógica detrás de esta terapia se basa en inhibir las moléculas que frenan la activación de los linfocitos T, células efectoras claves en la respuesta inmunológica anti-tumoral. Brevemente, los linfocitos T deben recibir dos señales para activarse correctamente, la primera involucra el reconocimiento del antígeno complementario a su receptor T (TCR) en el contexto del complejo mayor de histocompatibilidad (CMH) y la segunda implica la activación de moléculas co-estimuladoras, particularmente CD28 que interacciona con sus ligandos B7-1 (CD80) y B7-2 (CD86), que se hallan en las células presentadoras de antígenos o células blancos. Sin embargo, los linfocitos T no solo presentan en su superficie moléculas co-estimuladoras, sino también moléculas co-inhibitorias. Estas últimas, al activarse, inhiben las señales río abajo del TCR, atenuando la activación del linfocito T. Por esta razón se conoce a las moléculas co-inhibitorias como “puntos de control inmunológico” ya que son fundamentales para promover la homeostasis del sistema inmunológico y evitar así fenómenos de autoinmunidad y daño tisular2. A continuación describimos algunos puntos de chequeo inmunológico relevantes en el control de la respuesta antitumoral.
CTLA-4
En 1987 se clonó por primera vez la molécula co-inhibitoria CTLA-4 (del inglés Cytotoxic T-Lymphocyte Antigen 4). Esta molécula de la familia de las inmunoglobulinas se expresa como consecuencia de la activación del linfocito T y compite con el receptor CD28 por sus ligandos canónicos B7-1 y B7-2, con una afinidad 1000 veces mayor. La relevancia crítica de CTLA-4 en la homeostasis inmunológica fue comprobada utilizando modelos de ratones knockout en los cuales la ablación del gen de CTLA-4 genera un desorden linfoproliferativo sistémico que deviene en la muerte dentro de las 2-3 semanas de vida. Además de los linfocitos T CD4 y CD8 efectores, CTLA-4 está presente en linfocitos T regulatorios. En estas células, CTLA-4 se expresa constitutivamente y, a diferencia de su acción inhibitoria en linfocitos T efectores, en linfocitos T regulatorios es fundamental para su actividad supresora2.
A raíz del papel inhibitorio de CTLA-4 en la activación de los linfocitos T, se propuso su bloqueo como una herramienta que podría mejorar la respuesta anti-tumoral. Efectivamente, su inhibición mediante anticuerpos monoclonales inhibió el crecimiento tumoral en modelos murinos de fibrosarcoma, carcinoma de colon, próstata, mama, ovario, melanoma y linfoma, entre otros. La utilización de anticuerpos monoclonales neutralizantes de CTLA-4 fue aprobada por la Food and Drug Administration (FDA) en el año 2011 para el tratamiento de melanoma metastásico. Si bien esta terapia fue exitosa solo en un 30% de los pacientes, las respuestas obtenidas en aquellos individuos que sí respondieron fueron sumamente duraderas. De hecho, aproximadamente el 22% de los pacientes se mantuvieron libres de enfermedad por 10 años. Los efectos adversos de esta terapia se hallan relacionados con la activación incrementada de los linfocitos T, por lo que algunos pacientes pueden experimentar eccemas, irritación intestinal y endocrinopatías, síntoma de la inflamación exacerbada y daño tisular mediado por la respuesta inmunológica autorreactiva. Sin embargo, estos efectos adversos, a diferencia de una enfermedad autoinmune clásica, pueden revertirse con el cese de la terapia y controlarse con la administración de corticoides u otros agentes inmunomodulatorios.
PD-1 / PD-L1
Un punto de chequeo inhibitorio capaz de promover homeostasis inmunológica y limitar la respuesta protectiva y antitumoral se halla representado por el eje PD-1/PD-L1. A principios de la década del 90, el investigador japonés Tasuku Honjo identificó el receptor PD-1 y su participación en el proceso de muerte celular programada en los linfocitos T (de allí su nombre, Programmed Cell Death-1). Años más tarde, se logró desarrollar ratones deficientes en el gen codificante de PD-1 y se descubrió que estos animales sufrían procesos de autoinmunidad similares al lupus eritematoso sistémico, dando lugar al estudio de esta proteína como regulador de la homeostasis del sistema inmunológico. En el año 2000, en colaboración con el laboratorio del Dr. Honjo, los investigadores Arlene Sharpe y Gordon Freeman lograron cerrar el círculo al identificar los dos ligandos de PD-1: PD-L1 y PD-L2.
De esta forma, no solo confirmaron el rol inmunosupresor de esta vía de señalización, sino que además descubrieron la expresión de PD-L1 y PD-L2 en células tumorales, proponiendo así a PD-1 y PD-L1/L2 como mecanismos de escape tumoral. Este descubrimiento fue corroborado por el grupo de Lieping Chen, quien además introdujo por primera vez el bloqueo de PD-L1 mediante un anticuerpo monoclonal neutralizante, proponiéndolo como posible terapia para favorecer el rechazo del tumor. El receptor, PD-1 se expresa inmediatamente después de la activación de la vía del TCR silenciando señales de activación y proliferación3.
El bloqueo de CTLA-4 fue el puntapié inicial que demostró el poder antitumoral de la inmunoterapia por inhibición de moléculas co-inhibitorias; sin embargo, hoy en día su uso es más limitado que el bloqueo de PD-1. Por lo tanto, y dado que el bloqueo de PD-1 permitió la inhibición de la progresión tumoral en numerosos tipos de cáncer, se convirtió en la inmunoterapia por excelencia, no solo como monoterapia sino también en combinación con otras estrategias antitumorales. En 2014 se aprobó por primera vez su uso en pacientes con melanoma avanzado. Un año después, gracias a la excelente respuesta en casos de melanoma, fue aprobado para el tratamiento de cáncer de pulmón avanzado de células no pequeñas y carcinoma renal metastásico, en 2016 comenzó a utilizarse en cáncer de cabeza y cuello y linfoma Hodgkin y en 2017 se sumaron el carcinoma urotelial y todos aquellos tumores sólidos que presenten inestabilidad de microsatélite o errores en la maquinaria de reparación del ADN. Por otro lado, el bloqueo de PD-L1 fue aprobado en 2016 para el tratamiento de tumores uroteliales y de vejiga y ciertos tipos de cáncer de pulmón.
Se observó que los efectos adversos de estas terapias eran menores a los observados con el tratamiento con anticuerpos anti-CTLA-4. La explicación subyacente de este fenómeno radica en que CTLA-4 es fundamental para regular la activación de los linfocitos T en ganglios linfáticos, mientras que PD-1 actúa fundamentalmente en la periferia, una vez que los linfocitos T activados migran al sitio del tumor para reconocer por segunda vez al antígeno tumoral y eliminar a la célula blanco. Por este motivo es que el bloqueo de CTLA-4 favorece aún más la probabilidad de generación de clones autorreactivos3
La nueva generación
El repertorio de moléculas co-inhibitorias es cada vez más extenso. En vista de este complejo escenario, comenzaron a desarrollarse estrategias de bloqueo contra otros puntos de chequeo. Si bien hasta el momento no han sido aprobadas por las agencias regulatorias internacionales, algunas de estas terapias están generando muy buenos resultados en ensayos clínicos.
Tim-3
En los últimos años se demostró la expresión del receptor Tim-3 (del inglés: T cell immunoglobulin and mucin domain 3) en linfocitos infiltrantes de tumores o en linfocitos circulantes en diferentes tipos de cáncer incluidos cáncer de pulmón, cáncer de cabeza y cuello, carcinoma renal, cáncer de estómago y esófago, cáncer de próstata, cáncer de hígado, colorrectal, de ovario y linfoma no Hodgkin. En modelos animales, el bloqueo con anticuerpos neutralizantes mostró resultados diversos, ya que su capacidad para controlar el crecimiento tumoral no fue eficiente; sin embargo, su combinación con el bloqueo de PD-1 o CTLA-4 generó potenciación de la respuesta inmunológica anti-tumoral e inhibición del crecimiento del tumor4. Esta evidencia fue suficiente para comenzar los ensayos clínicos en pacientes. Actualmente se están llevando a cabo ensayos clínicos en Fase Ib-II evaluando la seguridad y eficacia de anticuerpos monoclonales contra Tim 3, solos o en combinación con el bloqueo de PD-1.
Lag-3
Lag-3 (del inglés: lymphocyte-activation gene-3) es una proteína transmembrana estructuralmente homóloga a CD4, presente en linfocitos T CD4, CD8 y células NK. Su mecanismo de acción es muy similar a otros puntos de control inhibitorios, ya que se expresa una vez activado el linfocito T y una vez que contacta con su ligando, inhibe las señales de proliferación y producción de citoquinas. El ligando conocido de Lag-3 es el CMH II, que a su vez es el ligando canónico de CD4. En este sentido, Lag-3 es capaz de competir con CD4 por el CMH II, y de la misma forma que CTLA-4 compite con CD28 por B7.1 y B7.2, lo hace con una afinidad mucho mayor5. El bloqueo in vivo de Lag-3 en modelos animales demostró la importancia de este receptor en la exhaustación de los linfocitos T específicos del tumor, pero no fue suficiente para controlar la progresión tumoral. Actualmente, cuatro ensayos de fase I estudian la aplicabilidad del bloqueo de Lag-3 en tumores sólidos y hematológicos, ya sea mediante anticuerpos monoclonales o la administración de Lag-3 soluble.
TIGIT / CD73
TIGIT (del inglés: T cell immunoglobulin and ITIM domain) posee una expresión restringida a linfocitos T, mayormente CD4 efectores y regulatorios, CD8 y NK; su cinética de expresión se correlaciona con la activación del linfocito T. A diferencia de CTLA-4, los ratones deficientes en esta proteína no presentan patologías inflamatorias o autoinmunes a menos que sean desafiados con algún antígeno propio. En modelos preclínicos, la inhibición de TIGIT una vez que el tumor se ha establecido no ha sido exitosa; sin embargo, los ratones deficientes en este receptor mostraron un menor crecimiento tumoral6. Actualmente, existe un ensayo clínico de Fase I en el que se estudia la seguridad y eficacia de un anticuerpo neutralizante de TIGIT en tumores sólidos avanzados.
La expresión de CD73 en el tumor inhibe la respuesta anti-tumoral. CD73 es una ecto-5´-nucleotidasa, cuya función es degradar AMP (generado a través de la acción de CD39 sobre ATP) a adenosina, cuya acumulación en el microambiente tumoral desencadena señales inhibitorias en los linfocitos T. De manera consistente, el bloqueo de CD73 en modelos animales inhibió el crecimiento tumoral y aumentó la eficacia de la transferencia adoptiva de linfocitos T. Se están llevando a cabo estudios clínicos de Fase I a fin de evaluar un nuevo anticuerpo monoclonal dirigido contra esta molécula7.
VISTA/IDO
VISTA (del inglés: V-domain Ig-containing suppressor of T-cell activation) surgió como un nuevo punto de control inhibitorio en los últimos 4 años. Su estructura es muy similar a PD-L1, aunque su ligando aún no ha sido descripto. Este receptor se expresa en linfocitos y con mayor densidad en células presentadoras de antígeno. En el microambiente tumoral, si bien su expresión varía con el tipo de tumor, solo se ha encontrado esta molécula en leucocitos infiltrantes, estando ausente en células tumorales, y co-localizando con CD11b, marcador de células mieloides8. En modelos preclínicos en los cuales se bloqueó VISTA pudo evidenciarse la función de esta molécula en el escape tumoral, observación que condujo al desarrollo de ensayos clínicos Fase I.
La enzima indoleamine-2,3-dioxygenase (IDO) es expresada por células de origen mieloide y células tumorales. Esta enzima cataboliza el triptófano del medio extracelular, generando la depleción de este aminoácido esencial y dando origen al metabolito inmunosupresor kinurenina. La eliminación del triptófano inhibe señales de activación y proliferación del linfocito T, ya que este aminoácido es esencial para su actividad, por lo que la célula se torna anérgica y se reprime en consecuencia, la respuesta anti-tumoral9. Se han desarrollado numerosos inhibidores en ensayos preclínicos, los cuales no tuvieron buenos resultados como monoterapia, pero son prometedores como combinación con el bloqueo de otros puntos de control inmunológicos o vacunas tumorales. De hecho, numerosos ensayos clínicos en Fases I, II y III se hallan actualmente activos. Particularmente, en junio de 2018 se presentó el resultado de un ensayo clínico en melanoma en el que no se encontró ventaja clínica de la combinación del bloqueo de IDO y anti-PD-1 por sobre la monoterapia con anticuerpos anti-PD-110.
Galectinas. Posibles blancos terapéuticos
Hasta el momento hemos descrito la actividad de receptores que al contactar con sus ligandos proteicos desencadenan señales inhibitorias sobre la activación y proliferación de células T efectoras. Sin embargo, existen vías alternativas de regulación que se basan en el reconocimiento entre proteínas y glicanos. Los glicanos forman parte de glicoproteínas y glicolípidos, presentes en la membrana celular y en la matriz extracelular. La combinación de las distintas estructuras de glicanos presentes en receptores de la superficie celular influyen sobre el destino de la misma, ya sea su activación, diferenciación, migración e incluso su muerte11. Este proceso de reconocimiento es mediado por proteínas endógenas solubles llamadas galectinas. El rol de las galectinas en la progresión y el escape tumoral ha sido establecido en numerosas publicaciones y validado en distintos tipos de cáncer como melanoma12, linfoma Hodgkin13, neuroblastoma14, cáncer de mama15, pulmón16 y páncreas17, 18. De acuerdo con su estructura, los miembros de la familia de galectinas pueden clasificarse en: a) “proto-tipo”, cuyo exponente con mayor relevancia en el escape tumoral es galectina-1; b) “tipo-tándem”, con galectina-9 como ejemplo más relevante en inmuno-oncología y c) “tipo-quimera”, representada por galectina-3, la cual ejerce un impacto importante sobre la progresión tumoral19. Del mismo modo que los puntos de control inhibitorios citados previamente, las galectinas son capaces de suprimir la respuesta inmunológica utilizando distintas estrategias. Galectina-1 se une a estructuras glicosídicas presentes en los receptores CD43 y CD45 del linfocito T, promoviendo su apoptosis11. Además, galectina-1 y -9 son capaces de generar la muerte selectiva de linfocitos Th1 y Th17, promoviendo la resolución de patologías autoinmunes, pero favoreciendo también el escape tumoral20, 21. Por otro lado, se ha documentado la unión de galectina-3 a CTLA-4 y LAG-3 como mecanismo de inmunosupresión de linfocitos T efectores20. Más aún, demostramos que galectina-1 es capaz de unirse a células dendríticas favoreciendo su capacidad tolerogénica22. A su vez, galectina-1 y -3 favorecen la diferenciación de macrófagos asociados a tumores hacia un fenotipo M2 anti-inflamatorio y junto con galectina-9, promueven el reclutamiento de células mieloides supresoras21. El interés que cobraron estas proteínas de unión a glicanos como nuevos blancos terapéuticos, no solo se debe a su naturaleza como “checkpoints”, sino también a sus efectos independientes del sistema inmunológico, tales como angiogénesis23 y metástasis15. En este sentido, observamos que galectina-1, a través de su unión a N-glicanos complejos presentes en el receptor tipo-2 del factor crecimiento del endotelio vascular (VEGFR2), promueve resistencia a las terapias anti-angiogénicas convencionales24. De esta manera, el bloqueo conjunto de galectina-1 y el factor de crecimiento vascular endotelial (VEGF) mostró tener un efecto sinérgico antitumoral, contrarrestando la resistencia adquirida a la terapia anti- VEGF. Dado el papel protagónico de estas galectinas en la progresión del tumor y la inhibición de la respuesta anti-tumoral, su bloqueo mediante anticuerpos neutralizantes emerge como una posible estrategia terapéutica para enriquecer el panorama de los tratamientos actuales.
Mecanismos de resistencia
Uno de los interrogantes que se generaron luego del advenimiento de las inmunoterapias fue cómo distinguir pacientes sensibles de resistentes a dichos tratamientos. Para responder esta cuestión es necesario por un lado determinar qué pacientes expresan los receptores/ ligandos que el tratamiento bloquea (PD-1/PD-L1, por ejemplo) y estudiar por otro lado, si el tumor presenta otros mecanismos que puedan compensar la señal inhibida por la terapia. En consecuencia, numerosos esfuerzos se hallan enfocados en explorar biomarcadores que puedan predecir o indicar el éxito de la terapia25.
En relación con el tratamiento anti-CTLA-4, se observó que el aumento de linfocitos, en conjunción con la expresión de moléculas co-estimuladoras y la producción de IFN-γluego del tratamiento se correlaciona con el beneficio clínico de estas terapias. En el caso de la terapia anti-PD-1, se observó que altos niveles de expresión de PD-L1 en el tejido tumoral se asocian con una mayor probabilidad de respuesta. Sin embargo, pacientes cuyos tumores no expresaban PD-L1 fueron capaces de responder al bloqueo de este receptor, por lo que aún no se lo considera un marcador predictivo ideal25.
Otro factor que ha cobrado relevancia como posible marcador predictivo es la cantidad de neo-antígenos que expresa cada tumor. Un neo-antígeno se define como una nueva proteína generada a raíz de una mutación exclusiva de la célula tumoral, por lo que se reconoce como extraña por el sistema inmunológico del huésped. A mayor número de neo-antígenos o mutaciones somáticas presentes en el tumor, mayor será la probabilidad de activar linfocitos que reconozcan y eliminen a las células que los expresan26.
Dado que el blanco principal de las inmunoterapias son los linfocitos T (directa o indirectamente), la cantidad de infiltrado CD3+CD8+ podría ser un posible biomarcador predictivo de la respuesta inmunológica de un paciente y de la capacidad de migrar al sitio tumoral para ejercer su función citotóxica. En este sentido, en cáncer colorrectal se describió por primera vez el score inmunológico, el cual se basa en la evaluación inmunohistoquímica de marcadores fenotípicos inmunológicos en el tejido tumoral, evaluando en particular la cantidad y localización de linfocitos T CD3+CD8+. A mayor cantidad de infiltrado en los márgenes y centro del tumor, mayor sería el índice del score, el cual, en numerosos tipos tumorales correlaciona con el éxito de la inmunoterapia. Como era previsible, se encontraron diversos mecanismos moleculares que inhiben la extravasación y migración de los linfocitos T activados desde ganglios linfáticos hacia tumores. Finalmente, la resistencia a inmunoterapia involucra una batería de células inmunosupresoras que son utilizadas por el tumor para inhibir la respuesta inmunológica efectora, tales como células mieloides supresoras, macrófagos M2 y neutrófilos asociados a tumores, células dendríticas tolerogénicas y linfocitos T regulatorios, entre otros. Estas células generan circuitos tolerogénicos que impiden el éxito de estrategias de inmunoterapias a través del bloqueo de puntos de control inmunológico.
Terapias basadas en anticuerpos agonistas de moléculas co-estimuladoras
Las terapias de bloqueo de puntos de control inmunológico, como PD-1 y CTLA-4, tienen como objeto liberar a la respuesta anti-tumoral de los frenos a la activación y proliferación de linfocitos. Las estrategias de inmunoestimulación, en cambio, tienen como blanco terapéutico a las moléculas co-estimuladoras27. En este sentido, existen moléculas co-estimuladoras que se expresan constitutivamente en células T naïve, tales como CD28 y CD27 mientras que otros receptores co-estimuladores de superficie como ICOS, CD137, OX40 y GITR, entre otros28, solo están presentes en la membrana una vez que la célula T se ha activado.
OX40 es un miembro de la superfamilia del receptor del factor de necrosis tumoral (TNFR) que posee importantes funciones co-estimuladoras29. Su ligando OX40L (CD252), se expresa en células presentadoras de antígeno. Tras la co-estimulación de linfocitos T por OX40, se activan vías intracitoplasmáticas asociadas a la activación y proliferación de los mismos, al mismo tiempo que se activan señales anti-apoptóticas promoviendo la memoria inmunológica y la actividad antitumoral. En estudios preclínicos en ratón se ha observado que la administración de agonistas de OX40 conduce a respuestas terapéuticas en distintos modelos tumorales como el modelo de cáncer de mama 4T1, el modelo de melanoma B16, el carcinoma de pulmón de Lewis y varios sarcomas inducidos químicamente30.
La capacidad de los agonistas de OX40 para sintonizar las respuestas inmunológicas, así como la expresión de OX40 en los linfocitos T CD4 y CD8 intratumorales y asociados a ganglios linfáticos que drenan tumores, condujo a examinar la estimulación de OX40 como un posible tratamiento para el cáncer. Recientemente, el uso de la monoterapia basada en anticuerpos monoclonales específicos de OX40 fue probado en un ensayo clínico de Fase 1 en pacientes con tumores sólidos, con resultados prometedores. Doce de cada 30 pacientes que recibieron un agonista de OX40 tuvieron regresión de al menos una lesión metastásica con solo un ciclo de tratamiento, con toxicidades menores que el tratamiento con anticuerpos anti-CTLA-4.
ICOS
ICOS (CD278) es una molécula coestimuladora inducible expresada principalmente en células T CD4 activadas. Se une a un solo receptor expresado por células dendríticas, células B y macrófagos denominado ICOSL o B7h. Su función coestimuladora favorece la proliferación de células T y la secreción de citoquinas. Sin embargo, los efectos de ICOS sobre la coestimulación de las células T parecen ser menos potentes que los ejercidos por CD28, probablemente porque no induce la producción de IL-2. Además, ICOS favorece la diferenciación funcional de linfocitos T helper foliculares, Th2 y Th17. En modelos preclínicos de tumores trasplantados, los agonistas estimulantes de ICOS no han demostrado ejercer una acción inmunoterapéutica potente. Sin embargo, se observó un aumento de ICOS en células T CD4 luego del tratamiento bloqueante con anti CTLA-4. Estudios preclínicos en modelos de melanoma y de cáncer de páncreas han demostrado efectos sinérgicos cuando se combinan la estimulación de ICOS con la inhibición de CTLA-431.
En la actualidad numerosas investigaciones se centran en el estudio de anticuerpos monoclonales agonistas de ICOS32. En este sentido, se ha desarrollado un anticuerpo monoclonal agonista humanizado, JTX-2011, que reconoce ICOS con potencial actividad antineoplásica. Tras la administración, el anticuerpo monoclonal agonista JTX- 2011 dirigido hacia ICOS une la molécula ICOS expresada en células T. Este fenómeno estimula la señalización mediada por ICOS, induce la proliferación de células T ICOS+, aumenta la supervivencia de linfocitos T citotóxicos (CTL) y potencia la respuesta inmune mediada por CTL contra células tumorales. Existen numerosos ensayos clínicos que evalúan los efectos del anticuerpo monoclonal agonista de ICOS como monoterapia y en combinación con terapias como anti CTLA-4 y anti PD-1.
GITR
La proteína relacionada con el receptor de TNF inducida por glucocorticoides, GITR (también conocida como TNFRSF18), pertenece a la familia de receptores de TNF. Se descubrió en hibridomas de células T murinas tratados con dexametasona: de allí así su nombre GITR. Se expresa en bajos niveles en células T CD4 y CD8, se regula hasta 24-72 h luego de la señalización del TCR y permanece expresada en la superficie del linfocito durante varios días. Las células T regulatorias expresan constitutivamente GITR; sin embargo, su función en estas células es aún controversial. Además, se ha observado que células NK, eosinófilos, basófilos, macrófagos y linfocitos B son capaces de expresar GITR. El ligando de GITR (GITRL), como en el caso con OX40L, se expresa en células presentadoras de antígeno activadas y en células endoteliales. Tras la unión de GITR a GITRL, la señalización conduce a la activación de la vía de proteínas quinasas activadas por mitógenos (MAPK). La coestimulación mediada por GITR en última instancia, aumenta la proliferación de células T y las funciones efectoras debido al incremento de IL-2 e IFN-γ. La unión GITR-GITRL protege a las células T de la muerte celular inducida por activación, dando lugar a un incremento de células T de memoria.
Se han observado efectos antitumorales luego de la estimulación de GITR en diferentes modelos experimentales utilizando un anticuerpo agonista anti-GITR en ratón (DTA-1)33. El tratamiento con el anticuerpo agonista DTA-1 demostró ser eficaz en modelos preclínicos de sarcoma, cáncer colorrectal y melanoma. Además, los agonistas de GITR han demostrado un efecto sinérgico antitumoral cuando se combinan con vacunas y con anticuerpos monoclonales como anti CTLA-4. Diferentes mecanismos podrían contribuir a los efectos antitumorales de GITR. Tal vez el mejor definido es la acción de agonistas de GITR en la inhibición de fenómenos mediados por células T regulatorias. Algunos estudios han demostrado que los agonistas de GITR aumentan la función de células T efectoras en el microambiente tumoral alterando la expresión de Foxp3 en células T regulatorias y anulando en consecuencia la función supresora de las mismas. Un anticuerpo agonista humanizado anti-GITR mAb humano, TRX518, ha sido desarrollado permitiendo la generación de un ensayo clínico de Fase I.
CD137
Una de las estrategias con aplicaciones clínicas más prometedoras es la que tiene como blanco terapéutico a la molécula CD137, un miembro de la superfamilia co-estimuladora de TNF que define el grado de activación, función y supervivencia de las células T. Originalmente el gen de CD137 (también llamado 41BB) se clonó en 1989 como un gen inducible de células T activadas y se demostró que la estimulación mediada por CD137 suprime el crecimiento tumoral en modelos murinos de sarcoma, mastocitoma y glioblastoma. Su expresión en células dendríticas, células NK y células T activadas, junto con su capacidad única de potenciar respuestas antitumorales, ha establecido desde entonces un papel importante para esta molécula como blanco atractivo para inmunoterapia contra el cáncer34. En efecto, CD137 se expresa en linfocitos T activados, pero no en reposo, y ha sido postulado como marcador de activación capaz también de discriminar células T CD4 y CD8 vírgenes específicas de antígeno y de memoria. Por otro lado, CD137 se halla constitutivamente expresada por células T regulatorias. Su ligando (CD137L) se expresa predominantemente por activación de células presentadoras de antígenos incluyendo células dendríticas, macrófagos y linfocitos B. Dos anticuerpos monoclonales anti-CD137 han sido desarrollados y aplicados a ensayos clínicos: el urelumab, IgG humanizada y el PF-05082566, una IgG2 humanizada. El estudio de Fase I del urelumab se inició en 2005 en una población de pacientes con melanoma, carcinoma de células renales, cáncer de ovario y próstata. Se observaron 3 respuestas completas y 4 casos de estabilización de la enfermedad. Sobre la base de la eficacia observada se inició un estudio de Fase II, el cual debió ser terminado debido a una alta incidencia de toxicidad hepática. En 2012, urelumab reingresó a la clínica después de que se demostrara que una dosis significativamente más baja podría alcanzar actividad terapéutica sin la limitante toxicidad. Urelumab está siendo actualmente evaluado en múltiples regímenes combinatorios: con cetuximab (anti-EGFR) en cáncer de cabeza y cuello y colorrectal, con rituximab (anti-CD20) en linfoma no Hodgkin de células B (NHL), y con elotuzumab (anti-CS1) en mieloma múltiple. El compuesto anti-CD137, PF-05082566, se encuentra actualmente en tres ensayos clínicos y los resultados de monoterapia evaluados en Fase I demostraron que la mejor respuesta clínica fue la estabilización de la enfermedad en 22% de los pacientes tratados sin inducir ninguna toxicidad. PF- 05082566 se está probando actualmente en combinación con rituximab y mogamulizumab (anti-CCR4).
Transferencia adoptiva de células inmunológicas como terapia contra el cáncer
La utilización de células autólogas o propias de un paciente para combatir un tumor ha cobrado gran importancia en los últimos años. No obstante, las primeras observaciones señalando que células del sistema inmunológico eran capaces de reconocer y destruir un tumor se remontan a la década del 50. Trabajos pioneros realizados por Mitchinson y Dube en ratones demostraron que la transferencia de células presentes en los ganglios linfáticos asociados al tumor era capaz de proteger del desarrollo tumoral y favorecer la eliminación de sarcomas establecidos35. Estos estudios fueron los responsables de impulsar la transferencia adoptiva de células como posible inmunoterapia en cáncer.
Transferencia adoptiva de linfocitos T
Poco se sabía acerca de la función de los linfocitos T hasta que en la década del 60 se demostró que estas células mediaban el rechazo de aloinjertos en modelos animales. Sin embargo, la transferencia de linfocitos T autólogos activados para el tratamiento contra el cáncer fue utilizada con éxito en melanoma recién en los años 90. En la actualidad su uso se ha extendido a otros tipos tumorales tanto sólidos como líquidos. Este tipo de terapia explota la actividad citotóxica y específica de linfocitos T para lograr eliminar las células tumorales. Se pueden diferenciar tres tipos de estrategias al abordar las transferencias adoptivas de células T: Células T antitumorales naturales; Células T con TCR transgénico (TCRt); y Células T con receptor quimérico de antígeno (T-CAR).
Células T antitumorales naturales
La presencia de linfocitos en el microambiente tumoral no siempre es indicadora de una respuesta antitumoral efectiva. Muchas veces su escasa abundancia coexiste con distintos puntos de chequeo inmunológicos y moléculas inmunosupresoras que actúan en forma coordinada para inhibir la reactivación in situ de los mismos, limitando así su expansión y función antitumoral. Hacia finales del siglo pasado fue posible purificar y expandir linfocitos T provenientes del tumor a los fines de reforzar su función efectora y reactivar de este modo la respuesta antitumoral. Metodológicamente es necesario purificar los linfocitos T infiltrantes del tumor (LiT) y expandirlos in vitro en presencia de IL-236,37. De esta manera, tras 5-6 semanas se pueden obtener hasta 1×1011 linfocitos capaces de ser transferidos. La activación en cultivo de estas células potencia su capacidad antitumoral al estar ausentes las moléculas inmunosupresoras presentes en el microambiente tumoral. Sin embargo, las respuestas objetivas en los primeros intentos fueron de corta duración, y los linfocitos transfundidos rara vez pudieron ser encontrados en circulación38. Un cambio crítico en la aplicación de esta terapia fue la linfodepleción no mieloablativa con ciclofosfamida y/o fludarabina previa a la transferencia adoptiva, la cual elimina los linfocitos presentes en circulación incluyendo células T regulatorias inmunosupresoras, generando así un nicho homeostático permisivo para el mantenimiento de los linfocitos T transferidos39. La administración de IL-2 concomitante asegura la sobrevida y autorrenovación de los linfocitos transferidos. La linfodepleción ha resultado fundamental para asegurar la efectividad de las terapias celulares con linfocitos y su utilización ha sido extendida a la transferencia de linfocitos con TCR transgénico o aquéllos con receptor quimérico de antígeno.
La utilización de LiT para el tratamiento de melanoma metastásico ha generado importantes beneficios clínicos con respuestas objetivas entre el 40-70% y remisiones completas duraderas40,41. No obstante, la utilización de linfocitos T antitumorales naturales tiene sus limitaciones. El tumor debe estar disponible para ser removido o biopsiado. Además, depende de la presencia de linfocitos T en el microambiente tumoral y de la frecuencia y diversidad de clones T reactivos contra antígenos tumorales. La dificultad para expandir eficientemente linfocitos antitumorales en cultivo también representa una barrera. Se estima que en el 50% de los casos es posible obtener linfocitos antitumorales. Esta falta de reproducibilidad restringe a muchos pacientes de poder acceder a esta terapia celular.
Antígenos tumorales como blancos terapéuticos
La identificación de antígenos tumorales como blancos terapéuticos ha revolucionado las terapias celulares posibilitó la personalización de los mismos. La selección del antígeno blanco es sumamente crítica en este sentido. Proteínas no mutadas, expresadas en forma incrementada como gp100, MART-1 o tirosinasa, o bien expresadas en manera aberrante como MAGE-A1, MAGE-A3, pertenecientes a la familia de los antígenos de cáncer/testículo, suelen ser buenos blancos terapéuticos a pesar de no ser completamente específicos. La secuenciación del exoma y transcriptoma a partir de pequeñas biopsias mejoró la comprensión de los mecanismos que operan durante la evolución tumoral, pudiendo distinguir mutaciones driver (blancos de las terapias convencionales) y predecir neoantígenos tumorales. La transferencia adoptiva de linfocitos con TCR transgénicos o de linfocitos con receptores quiméricos de antígenos para el tratamiento del cáncer explotan este conocimiento.
Linfocitos con TCR transgénico
Las tecnologías de secuenciación de la región determinante de la complementariedad del TCR han posibilitado la identificación de receptores específicos contra antígenos expresados en células tumorales. Curiosamente, muchos antígenos, como gp-100, MART-1 y CEA se hallan conservados entre tumores de distintos pacientes. La posibilidad de secuenciar un TCR específico contra un antígeno tumoral, clonarlo e introducirlo como transgén a linfocitos extraídos de sangre periférica permite extender el universo de pacientes tratables con inmunoterapia celular. En este procedimiento se basa la tecnología de linfocitos con TCR transgénicos. Esta estrategia hace prescindible la necesidad de aislar linfocitos T específicos del tumor, lo cual permite tratar pacientes con tumores inmunológicamente ignorantes o en los que se dificulte cultivar LiT. Se utilizan linfocitos purificados de la sangre del paciente que luego son transducidos con un vector retroviral o lentiviral conteniendo un cassette de expresión con las cadenas α y β del TCR de interés. De esta manera se obtiene un gran número de linfocitos T antitumorales capaces de ser reclutados al microambiente tumoral, reconocer al tumor, ejercer su acción citotóxica y proveer protección sostenida en el tiempo. No obstante, su beneficio está restringido a la compatibilidad HLA del paciente42.
El primer ensayo exitoso fue realizado en melanoma en 1999. Desde entonces, otros trabajos han reportado la transferencia exitosa de linfocitos con TCR transgénicos. En 2006 se realizó la primera fase clínica en melanoma con TCR transgénicos contra el antígeno MART-1. En 2011 se utilizó esta estrategia para dirigir linfocitos contra el antígeno NY-ESO-1 en sarcoma, obteniendo un beneficio clínico de 67% de respuestas objetivas43. No obstante, una serie de obstáculos suelen limitar la eficacia y el alcance de estas terapias.
Afinidad del TCR
El TCR suele presentar una baja afinidad y/o avidez por el CMH-péptido tumoral debido, mayormente, a los mecanismos de tolerancia central44. Se estima que esta afinidad es 1,5 veces menor, en escala logarítmica, que aquélla observada entre el TCR y el CMH-péptido viral. Se ha implementado mutagénesis racional para aumentar la afinidad del TCR por el CMH con antígeno tumoral. Otra alternativa ha sido la de inmunizar ratones que expresen CMH humanos en sus tejidos, con péptidos de tumores humanos. Los linfocitos T citotóxicos obtenidos de esta manera son aislados y su TCR es clonado e introducido en linfocitos T humanos. Este ha sido el caso de los antígenos tumorales gp100, p53, CEA y MAGE-A3. Si bien estas estrategias son capaces de proveer TCR con alta afinidad, esto no necesariamente implica un beneficio clínico. De hecho, una mayor afinidad puede resultar en el desarrollo de toxicidad sistémica. En ensayos clínicos en melanoma contra los antígenos gp-100 y MART-1, la utilización de TCR murinos resultó en la regresión del tumor en el 20% de los casos, junto con la destrucción de melanocitos sanos en la piel, oído y retina donde la expresión de los antígenos es menor que en el tumor45,46. Este tipo de toxicidad denominada ON target/OFF tumor que resulta del incremento de la afinidad puede ser tratada local- o sistémicamente con corticoides. Si bien el incremento de la afinidad puede mejorar la eficacia de la terapia, los potenciales efectos adversos deben ser monitoreados y tratados.
Expresión de CMH tumoral
La utilización de linfocitos T con TCR transgénico supone la reactivación de los mismos en el microambiente tumoral a través del contacto entre el TCR y el CMH-antígeno presente en la superficie de células tumorales. Sin embargo, el silenciamiento del CMH constituye un mecanismo de escape en un gran número de tipos tumorales y constituye una de las principales resistencias a la terapia celular con TCR transgénico. Por otra parte, la afinidad de las moléculas del CMH por sus péptidos hace que sea muy difícil dirigir la terapia contra antígenos de naturaleza lipídica y glicosídica, mucha veces más abundante y específica que la peptídica.
Linfocitos con receptor quimérico de antígeno
La baja afinidad de los TCR transgénicos, la potencial toxicidad de los TCR híbridos y la necesidad de antígenos restringidos a moléculas de CMH-péptido en la superficie de la célula tumoral impulsó el desarrollo de otro tipo de terapia celular. Hace varias décadas, el advenimiento de anticuerpos monoclonales facilitó la explosión de herramientas específicas y de alta afinidad contra moléculas tumorales. La revolución de la inmunoterapia de los últimos años ha capitalizado en este desarrollo pre-existente para la generación de receptores capaces de activar a linfocitos T con una alta especificidad. Los denominados receptores quiméricos de antígenos o Chimeric Antigen Receptors (CARs) constan de una porción extracelular compuesta por un scFv (single-chain variable fragment) derivado de un anticuerpo monoclonal dirigido contra una molécula presente en la superficie de la célula tumoral. Este scFv se encuentra fusionado a un dominio formado por las porciones intracelulares del CD3ζ del TCR y de moléculas co-estimuladoras tales como CD28 y/o CD137. De esta manera, al contactar con el antígeno, el linfocito recibe la primera y segunda señal de activación necesaria para montar una respuesta efectora. El diseño de los CARs ha ido evolucionando durante los últimos años. Inicialmente desarrollados por Gross y colaboradores en 1989, los denominados CARs de primera generación cuentan con el dominio CD3ζ, mientras que aquéllos de segunda y tercera generación contienen además uno o dos dominios de co-estimulación, respectivamente47,48. La incorporación de los dominios intracelulares de CD28 y CD137 mejoró notablemente la persistencia in vivo y la capacidad efectora de los linfocitos, comparado con los CARs de primera generación que al activarse en ausencia de una segunda señal generalmente sufrían fenómenos de anergia.
Una ventaja que presenta esta estrategia es la posibilidad de dirigir la especificidad contra moléculas proteicas, lipídicas y sacarídicas no restringidas al CMH, contribuyendo a la gran versatilidad de este tipo de inmunoterapia celular. El desarrollo de esta tecnología contra los antígenos CD19, CD20, CD22, CD30 y CD138 ha revolucionado el tratamiento de neoplasias hematológicas recurrentes como linfoma Hodgkin (LH) y no Hodgkin (LNH), leucemia linfoblástica aguda (LLA), leucemia linfocítica crónica B (LLC) y mieloma múltiple49. En estos casos, la quimioterapia suele ser el estándar de tratamiento, seguido de un transplante alogeneico de células madre hematopoyéticas, aunque para ello se debe alcanzar un estado clínico con enfermedad residual mínima negativa. La mayoría de los pacientes logra acceder a esta terapia, lo cual se refleja en un 85% y un 69% de sobrevida a 5 años en pacientes pediátricos y adultos, respectivamente. Sin embargo, aquellos pacientes con enfermedad resistente a la quimioterapia exhiben un muy pobre pronóstico. Los ensayos clínicos en estos pacientes, su mayoría con una supervivencia menor a los 6 meses, han suscitado regresiones dramáticas con un 90% de respuestas completas o enfermedad residual mínima negativa y con una tasa de sobrevida del 78%. Quizá el caso paradigmático que ilustra el éxito de esta terapia celular es el caso de la LLA utilizando un CAR contra la molécula CD19. Numerosos ensayos clínicos realizados en distintas instituciones con CARs de segunda y tercera generación han demostrado considerables beneficios clínicos, con respuestas completas alrededor del 90% y la posibilidad de recibir un transplante alogeneico de células madre hematopoyéticas50, 51. El éxito de los CARs en los ensayos clínicos para el tratamiento de leucemias ha motivado la aprobación de esta terapia por la Food and Drug Administration para su uso en la clínica, y lleva el nombre de tisagenlecleucel. El desarrollo de CARs dirigidos contra tumores sólidos no se encuentra tan avanzado como el de las neoplasias líquidas, lo cual refleja una de las limitaciones de este tipo de terapias que es la infiltración de los linfocitos al tumor. No obstante, existen ensayos clínicos de Fase I para neuroblastoma contra el antígeno GD2, Fase I/II para glioblastoma y gliomas malignos y contra EGFRvIII para sarcoma utilizando CARs anti-Her252.
La precisión con la cual las células T han sido diseñadas para reconocer el tumor ciertamente reduce el riesgo de efectos adversos generales como el observado en las quimioterapias convencionales. Sin embargo, numerosos estudios han reportado fenómenos de toxicidad con un rango de intensidad variable, mediado por las propias células inmunológicas transferidas53. Algunos ejemplos son: el síndrome de liberación de citoquinas (SLC) proinflamatorias como IL-1, IL-6, TNF e IFN-γ, provocado por la activación masiva de los linfocitos T que puede llevar a la caída de la presión arterial y complicaciones neurológicas54; el efecto ON target/OFF tumor, provocado por el reconocimiento del antígeno tumoral pero expresado por células normales (un ejemplo es el CD20, cuyo reconocimiento OFF Tumor genera aplasia de células B, sin poner en riesgo la vida del paciente); el efecto OFF Target/OFF Tumor, el cual puede ser evitado inhibiendo la expresión del TCR transgénico para garantizar que el único antígeno reconocido por ese linfocito sea aquel complementario al CAR. Finalmente, el síndrome de lisis del tumor (SLT), hace referencia a la combinación de complicaciones metabólicas que pueden llevar a la falla renal aguda, derivada de la liberación masiva de potasio, fosfato y ácidos nucleicos a causa de la muerte tumoral.
Mejorando la bioseguridad y función de células T-CAR
Las distintas complicaciones suscitadas por la actividad de las células T-CAR impulsó el desarrollo de nuevas metodologías con el fin de incrementar la bioseguridad de esta inmunoterapia celular55.
Toxicidad
La toxicidad asociada a la terapia con células T-CAR ha sido abordada de diferentes maneras.
Eliminación de células T-CAR
La incorporación del gen suicida codificante de la enzima timidina kinasa (TK) en las células T permite su rápida eliminación mediante la inyección de la droga ganciclovir55. La TK es capaz de convertir al ganciclovir en una sustancia bioactiva (ganciclovir fosforilado) que inhibe la incorporación de dGTP por la ADN polimerasa gatillando la apoptosis. Otro método para poder eliminar selectivamente las células genéticamente modificadas es programarlas para que expresen moléculas como CD20 o EGFR. De esta manera, las células pueden ser eliminadas mediante la inyección de los anticuerpos específicos rituximab y cetuximab, respectivamente.
Activación condicional
Una estrategia interesante para limitar los efectos ON target/OFF tumor es incrementar la especificidad de los linfocitos contra las células tumorales aumentando el número de antígenos que estos deben contactar para activarse. Una manera de lograrlo es disociando los dominios de activación del CD3ζ de los dominios de coestimulación CD28/CD137 en dos receptores quiméricos distintos. De esta manera, la activación completa de las células T-CAR ocurre únicamente al contactar con 2 antígenos presentes en las células tumorales. No obstante, un potencial problema de este sistema es la generación de anergia clonal por activación incompleta al contactar con un único antígeno.
Los denominados SynNotch CAR son células que expresan un receptor quimérico que al contactar al primer antígeno se produce la liberación del factor de transcripción Notch que luego induce la expresión del receptor quimérico que contiene los dominios CD3ζ y CD28/ CD13756.
Persistencia in vivo
Una de las principales razones que condicionan el éxito de la terapia con células T-CAR es la persistencia y expansión de las mismas in vivo. La administración de IL-2 junto con un protocolo de linfodepleción parecería promover la sobrevida de las células transferidas. Sin embargo, algunos trabajos reportaron que IL-2 favorece un fenotipo regulatorio57. Por otro lado, la suplementación con IL-7 e IL-15 ha mostrado ser importante para la expansión y activación de células T al favorecer la generación de células de memoria y restringir la diferenciación de células T regulatoria58,59. La IL-15, además, parecería incrementar la actividad de la telomerasa, evitando así fenómenos de senescencia.
La exhaustación clonal, la apoptosis inducida por activación y la baja tasa de autorrenovación atentan contra la persistencia de las células diferenciadas. En este aspecto, estudios recientes han señalado que la eficacia terapéutica de células T-CAR varía de acuerdo a la composición de subpoblaciones T pre-existentes en el contexto de las células inoculadas60. Distintos autores demostraron que las poblaciones T de memoria stem CD45RA+ CD45RO+ CD95+ CCR7+ en mayor medida que las T de memoria central CD45RA– CD45RO+ CCR7+ y más aún que las de memoria efectora CD45RA– CD45RO+ CCR7–, inducen una mayor respuesta antitumoral y confieren una memoria inmunológica superior. La composición de células citotóxicas (CD8) y helper (CD4) parecería también modular la magnitud de la respuesta antitumoral. Particularmente para el caso de LLC CD19+, se ha observado un sinergismo al transferir células CD4 y CD8 en una relación 1:1.
Resistencia a la inhibición
La exhaustación clonal de linfocitos T como respuesta a la exposición crónica a un antígeno es motivo de resistencia a distintas terapias antitumorales. La expresión inducida de PD-1, TIM-3 o LAG-3 en linfocitos T CD8 y CD4 limita su capacidad efectora. La presencia de linfocitos T exhaustos en el tumor es motivo de resistencia a diferentes terapias oncológicas. En este sentido, la incorporación de una variante dominante negativa o un short-hairpin ARN contra PD-1 limita la inhibición de las células T-CAR por exhaustación61.
Los denominados “CARs armados” son capaces de producir IL-12 en respuesta a la activación por un sistema dependiente del factor nuclear de activación T (NFAT), evitando así la inhibición local del microambiente tumoral62.
En resumen, la tecnología de células T-CAR es un área en constante desarrollo y promete generar grandes beneficios clínicos. Un gran avance ha tenido lugar en neoplasias hematológicas, aumentando el porcentaje de respuesta completas de pacientes con un mal pronóstico de sobrevida. La utilización de esta terapia para tumores sólidos es inminente y supone una revolución en su tratamiento si es abordada de manera racional, considerando aspectos claves como lo son la infiltración al tumor y el bloqueo de los mecanismos inmunosupresores y de los puntos de chequeo inmunológicos.
Combinaciones terapéuticas
Si bien las estrategias terapéuticas resumidas en este artículo han tenido éxito relativo en la clínica, la tasa de respuestas objetivas sigue estando por debajo de la deseada. En este contexto, el enfoque actual consiste en combinar las herramientas terapéuticas de manera racional a los fines de potenciar sus efectos particulares en cada evento de la respuesta inmune antitumoral63 (Fig. 1).
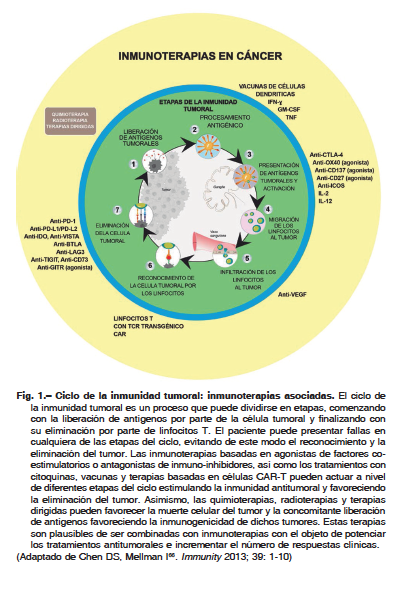
La combinación de anticuerpos bloqueantes de PD-1 y CTLA-4 ha sido aprobada en 2015 para melanoma, aunque no todos los pacientes responden eficientemente. Por otro lado, datos preclínicos demuestran gran porcentaje de respuestas completas cuando se utilizan terapias celulares. Estas últimas, en conjunto con agentes bloqueantes de los puntos de chequeo orientados a proteger la función de las células T transferidas al microambiente tumoral, están siendo evaluadas actualmente. Ensayos clínicos en LLA y LLC con células T-CAR contra CD19 de segunda y tercera generación en combinación con anticuerpos contra PD-1 y CTLA-4 han mostrado importantes beneficios clínicos64. Aún cuando el enfoque combinatorio puede incrementar la fracción de pacientes que respondan a la inmunoterapia, el tratamiento personalizado basado en marcadores predictivos tiene el potencial de mejorar aún más este porcentaje. De este modo, es prioridad establecer biomarcadores específicos que permitan monitorear la efectividad clínica de combinaciones de inmunoterapia, y delinear fenómenos de resistencia farmacológica25, 65.
Agradecimientos: El trabajo en nuestro laboratorio cuenta con el apoyo de subsidios de la Agencia Nacional de Promoción Científica y Tecnológica (PICT V 2014-3687; PICT I 2012- 2440), CONICET, Universidad de Buenos Aires, Fundación Sales y Fundación Bunge & Born.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Coley WB. The treatment of inoperable sarcoma by bacterial toxins (the mixed toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc R Soc Med. 1910; 3: 1-48.
2. Bayry J. Autoimmunity: CTLA-4: a key protein in autoimmunity. Nat Rev Rheumatol 2009; 5: 244-5.
3. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15: 486-99.
4. Sakuishi K, Apetoh L, Sullivan JM. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 2010; 207, 2187-94
5. Goldberg, MV, Drake CG. LAG-3 in cancer immunotherapy. Curr Top Microbiol Immunol 2011; 344: 269-78.
6. Manieri NA, Chiang EY, Grogan JL. TIGIT: A key inhibitor of the cancer immunity c. Trends Immunol 2017; 38: 20-8.
7. Antonioli L, Pacher P, Vizi ES, et al. CD39 and CD73 in immunity and inflammation. Trends Mol Med 2013; 19: 355-67.
8. Lines JL, Pantazi E, Mak J, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res 2014; 74: 1924-32.
9. Moon, YW, Hajjar, J, Hwu, P, et al. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J Immunother Cancer 2015; 3: 51.
10. Long GV, Dummer R, Hamid O, et al. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: Results of the phase 3 ECHO-301/KEYNOTE-252 study. Australia: ASCO, 2018.
11. Rabinovich GA, Croci DO. Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity 2012; 36: 322-35.
12. Rubinstein N, Alvarez M, Zwirner NW, et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection: A potential mechanism of tumor-immune privilege. Cancer Cell 2004; 5: 241-51.
13. Juszczynski P, Ouyang J, Monti S, et al. The AP1-dependent secretion of galectin-1 by Reed–Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc Nat Acad Sci USA 2007; 104: 13134-9.
14. Soldati R, Berger E, Zenclussen AC, et al. Neuroblastoma triggers an immunoevasive program involving galectin- 1-dependent modulation of T cell and dendritic cell compartments. Int J Cancer 2012; 131: 1131-41.
15. Dalotto-Moreno T, Croci DO, Cerliani JP, et al. Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res 2013: 73: 1107-17.
16. Banh A, Zhang J, Cao H, et al. Tumor galectin-1 mediates tumor growth and metastasis through regulation of T-cell apoptosis. Cancer Res 2011; 71: 4423-31.
17. Martínez-Bosch N, Fernández-Barrena MG, Moreno M, et al. Galectin-1 drives pancreatic carcinogenesis through stroma remodeling and Hedgehog signaling activation. Cancer Res 2014 74: 3512-24.
18. Orozco CA, Martinez-Bosch N, Guerrero PE, et al. Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor-stroma crosstalk. Proc Natl Acad Sci USA 2018; 115: E3769-78.
19. Rabinovich GA, Conejo-Garcia JR. Shaping the immune landscape in cancer by galectin-driven regulatory pathways. J Mol Biol 2016; 428: 3266-81.
20. Toscano MA, Bianco GA, Ilarregui JM, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol 2007; 8: 825-34.
21. Mendez-Huergo SP, Blidner AG, Rabinovich GA. Galectins: emerging regulatory checkpoints linking tumor immunity and angiogenesis. Curr Opin Immunol 2017; 45: 8-15.
22. Ilarregui JM, Croci DO, Bianco GA, et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1- driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat Immunol 2009; 10: 981-91.
23. Croci DO, Salatino M, Rubinstein N, et al. Disrupting galectin-1 interactions with N-glycans suppresses hypoxia-driven angiogenesis and tumorigenesis in Kaposi’s sarcoma. J Exp Med 2012; 209: 1985-2000.
24. Croci DO, Cerliani JP, Dalotto-Moreno T, et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 2014; 156:744-58.
25. Nishino M, Ramaiya NH, Hatabu H, et al. Monitoring immune- checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol 2017; 14: 655- 68.
26. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348: 69-74.
27. Peggs KS, Quezada SA, Allison JP. Cancer immunotherapy: co-stimulatory agonists and co-inhibitory antagonists. Clin Exp Immunol 2009; 157: 9-19.
28. Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004; 18: 676-84.
29. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of Co-stimulation in Cancer Immunotherapy Directed Against CD137, OX40, GITR, CD27, CD28, and ICOS. Sem Oncol 2015; 42: 640-55.
30. Linch SN, McNamara MJ, Redmond WL. OX40 Agonists and combination immunotherapy: Putting the pedal to the metal. Front Oncol 2015; 5: 34.
31. Fan X, Quezada SA, Sepulveda MA, et al. Engagement of the ICOS pathway markedly enhances efficacy of CTLA- 4 blockade in cancer immunotherapy. J Exp Med 2014; 211: 715-25.
32. Amatore F, Gorvel L, Olive D. Inducible Co-Stimulator (ICOS) as a potential therapeutic target for anti-cancer therapy. Expert Opin Ther Targets 2018; 22: 343-51.
33. Knee DA, Hewes B, Brogdon JL. Rationale for anti-GITR cancer immunotherapy. Eur J Cancer 2016; 67: 1-10.
34. Weigelin B, Bolaños E, Teijeira A, et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc Natl Acad Sci USA 2015; 112: 7551-6.
35. Mitchinson NA, Dube OL. Studies on the immunological response to foreign tumor transplants in the mouse. J Exp Med 1955; 102: 157-77.
36. Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 1976; 193: 1007-8.
37. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986; 233: 1318–21.
38. Rosenberg SA, Packard BS, Aebersold PM. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med 1988; 319: 1676-80.
39. Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298: 850-4.
40. Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011; 17 : 4550-7.
41. Besser MJ, Shapira-Frommer R, Itzhaki O, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res 2013; 19: 4792-800.
42. Klaver Y, Kunert A. Sleijfer S, et al. Adoptive T-cell therapy: A need for standard immune monitoring. Immunotherapy 2015; 7: 513-33.
43. Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 2011; 29: 917-24.
44. Nicholson E, Ghorashian S, Stauss H, et al. Improving TCR gene therapy for treatment of haematological malignancies. Adv Hematol 2012; ID: 404081.
45. Kawakami Y, Eliyahu S, Delgado CH, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA 1994; 91: 6458-62.
46. Kawakami Y, Eliyahu S, Delgado CH, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci USA 1994; 91: 3515-9.
47. Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004; 18: 676-84.
48. Song DG, Ye Q, Carpenito C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res 2011; 71: 4617-27.
49. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348: 62-8.
50. Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012; 119: 2709-20.
51. Grupp SA, Kalos M, Barrett D. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368: 1509-18.
52. Kakarla S, Gottschalk S. CAR T cells for solid tumors: armed and ready to go? Cancer J 2014; 2: 151-5.
53. Kalaitsidou M, Kueberuwa G, Schütt A, et al. CAR T-cell therapy: toxicity and the relevance of preclinical models. Immunotherapy 2015; 7: 487-97.
54. Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity 2013; 39: 49-60.
55. Bonifant CL, Jackson HJ, Brentjens RJ, et al. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics 2016; 3: 16011.
56. Roybal KT, Rupp LJ, Morsut L, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 2016; 164: 770-9.
57. Zhang H, Chua KS, Guimond M, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med 2005; 11: 1238-43.
58. Ku CC, Murakami M, Sakamoto A, et al. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science 2000; 288: 675-8.
59. Berger C, Berger M, Hackman RC, et al. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood 2009; 114: 2417-26.
60. Wang X, Popplewell LL, Wagner JR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood 2016; 127: 2980-90.
61. Liu X, Ranganathan R, Jiang S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second generation CAR T-Cells in advanced solid tumors. Cancer Res 2016; 76: 1578-90.
62. Chmielewski M, Kopecky C, Hombach AA, et al. IL‑12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res 2011; 71: 5697-706.
63. Swart M, Verbrugge I, Beltman J. Combination approaches with immune-checkpoint blockade in cancer therapy. Front Oncol 2016; 6: 223.
64. John LB, Devaud C, Duong CP, et al. Anti‑PD‑1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res 2013; 19 : 5636-46.
65. Sunblad V, Morosi LG, Geffner JR, et al. Galectin-1: a jack-of-all-trades in the resolution of acute and chronic inflammation. J Immunol 2017; 199: 3721-30.
66. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013; 39: 1-10.