
NÉLIDA RAIMONDO1, CRISTINA ECHEVERRÍA2, FERNANDO STENGEL3, GRACIELA PELLERANO4,
JENNIFER KREIMER5, 6, LUIS MAZZUOCCOLO7, MATÍAS MASKIN3, 4, NORA KOGAN5, 8,
PAULA LUNA9, SIMÓN GUSIS5
1Servicio de Dermatología, Hospital Aeronáutico Central, 2Servicio de Dermatología, Hospital General de Agudos Eva Perón, 3Clínica Buenos Aires Skin, 4Instituto Universitario Centro de Educación Médica e Investigaciones Clínicas (CEMIC), 5Hospital Ramos Mejía, 6Hospital Sirio Libanés, 7Servicio de Dermatología, Hospital Italiano de Buenos Aires, 8Fundación Centro de Neurociencias, Investigación y Tratamiento (CENIT), 9Hospital Alemán, Buenos Aires, Argentina
Resumen Con la aparición de los tratamientos biológicos, se ha modificado la terapéutica de muchas enfermedades, en especial las reumatológicas, dermatológicas y oncológicas. Debido al alto costo de estos productos y el vencimiento de las patentes, la industria farmacológica desarrolla los biosimilares, fármacos que son una versión (copia) de la sustancia de un medicamento biológico original, y que pueden facilitar el acceso a estos tratamientos. Son elaborados de acuerdo a exigencias específicas de organismos reguladores en cuanto a calidad, eficacia y seguridad, y debe demostrarse que son comparables al medicamento de referencia. Este trabajo revisa las normativas regulatorias internacionales y nacionales, las controversias que rodean a los biosimilares y presenta la posición de un grupo de expertos con respecto al uso de biosimilares.
Palabras clave: biológicos, biosimilares, consenso, psoriasis
Abstract Biosimilars: Expert consensus of the Latin American Society of Psoriasis (SOLAPSO) in Argentina. With the appearance of biological treatments, therapeutics has changed in many rheumatological, dermatological and oncological diseases. Due to the high cost of these biological medicaments and the expiration of patents, the pharmacological industry develops biosimilars, drugs that are a version (copy) of the substance of the original biological medicine, with the aim of facilitating access to these treatments. These biosimilars are prepared according to the specific requirements of regulatory bodies in terms of quality, efficacy and safety, and must be shown they are comparable to the reference product. This paper reviews the international and national regulatory framework, the controversies surrounding biosimilars, and presents the position of a group of experts regarding the use of biosimilars.
Key words: biological, biosimilar, consensus, psoriasis
Recibido: 5-III-2018 Aceptado: 21-VI-2018
Dirección postal: Cristina Echeverría, Cabello 3636 6° A, 1425 Buenos Aires, Argentina
e-mail: cmecheverria@gmail.com
La llegada e inclusión en la terapéutica de los medicamentos biológicos1, sustancias sintetizadas a partir de organismos vivos por medio de diferentes procesos, como tecnología de ADN recombinante, expresión controlada de genes o tecnologías de anticuerpos, tiene diversas implicancias para el cuerpo médico. Estos productos han cambiado en forma relevante el tratamiento de muchas enfermedades, como las inflamatorias y autoinmunes2 produciendo un control más efectivo de los síntomas y mejorando la calidad de vida, entre otros resultados3. Estos fármacos biológicos reciben aprobación regulatoria y están bajo protección de patentes por 20 años. Su aparición data a principios de los años 80 y actualmente representan uno de los sectores de más rápido crecimiento de la industria farmacéutica en todo el mundo.
Sin embargo, su acceso global es limitado en muchos países debido al costo elevado4 de estas terapias, lo que impacta en los sistemas sanitarios. Con la expiración de las patentes de algunos principios activos, surge el desarrollo de los biosimilares por parte de la industria biotecnológica. Un biosimilar (BS), es definido por la Agencia Europea del Medicamento (EMA) como “un producto médico biológico que contiene una versión (copia) de la sustancia de un medicamento biológico original autorizado”5. Este es producido de acuerdo a exigencias específicas de organismos reguladores en cuanto a calidad, eficacia y seguridad, y debe demostrar que es comparable al medicamento de referencia, una vez que su patente ha expirado. En septiembre de 2013, la EMA aprobó por primera vez a Remsima© e Inflectra© como fármacos BS de Remicade©6, 7. En 2018, fueron aprobados, también por la EMA, BS de dos importantes medicamentos oncológicos (trastuzumab y bevacizumab)8. Actualmente otros BS están en fases avanzadas de investigación. En Argentina, la ANMAT (Administración Nacional de Medicamentos, Alimentos y Tecnología Médica) define en su disposición 5755/96 a los BS como una especialidad medicinal o farmacéutica similar, a aquella que contiene el (los) mismo(s) principio(s) activo(s), la(s) misma(s) concentración(es), la(s) misma(s) forma(s) farmacéutica(s), la misma vía de administración, la misma indicación terapéutica, la misma posología, y que es equivalente al producto de referencia, pudiendo diferir en características tales como tamaño y forma, excipientes, período de vida útil y envase primario.
Las empresas productoras de biosimilares no tienen acceso al clon, ni a su banco de células originales, como tampoco a los procesos de fermentación y purificación originales o al principio activo que contiene el medicamento. Esto puede llevar, a que variaciones en las impurezas o en la proporción de las diferentes sustancias que contienen, pongan en duda la similitud con el original en lo que respecta a su seguridad y eficacia, y en consecuencia sobre los efectos que pudiese tener para la salud. En ello subyacen las controversias alrededor de los BS que serán analizadas en este trabajo, como extrapolación de indicaciones, inmunogenicidad, intercambiabilidad, nomenclatura y trazabilidad. La Sociedad Latinoamericana de Psoriasis (SOLAPSO) de Argentina, entidad que reúne a especialistas de nuestro país, establece y propone en este trabajo, pautas sobre el manejo y la seguridad en el uso de estos nuevos productos.
Proceso de desarrollo y aprobación de los biosimilares
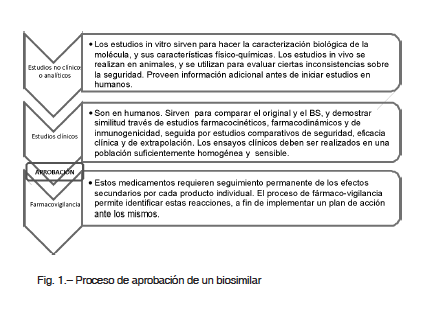
Para la aprobación de un BS se debe demostrar la alta similitud con el producto original. Debido a la complejidad de las moléculas biológicas y la falta de acceso a los datos de manufactura de los propietarios, para desarrollar un BS se diseña un proceso de ingeniería reversa del producto autorizado. Estos son aprobados sobre la base de una vía regulatoria diferente para genéricos y originales. Para ello, se debe demostrar comparabilidad con el producto de referencia en términos de calidad, características, actividad biológica, seguridad, inmunogenicidad y eficacia. Ello se logra a través de un ejercicio de comparabilidad paso a paso, de acuerdo a las guías EMA, que incluye estudios no clínicos y uno o más ensayos clínicos9 (Fig. 1). Por lo tanto, el objetivo de un programa de desarrollo de BS no es demostrar beneficio en el tratamiento del paciente, sino que los ensayos clínicos son diseñados para representar el modelo más sensible a fin de detectar diferencias10-12. Si el producto evaluado falla en la comparación en cualquiera de las etapas, no es autorizado como BS. Solamente las versiones de las copias que completan exitosamente el proceso, pueden ser llamados biosimilares13.
Regulaciones internacionales
El camino previsto por las regulaciones mundiales vigentes para la concesión de la autorización de comercialización de los BS, trata de minimizar los costos de desarrollo y acelerar su acceso al mercado, creando una
convergencia de intereses entre los servicios de salud, preocupados por la sostenibilidad, y los fabricantes. Por ello, entre 2003 y 2005, la EMA, ha sido la primera organización en abordar el proceso de producción y autorización de BS, a través de varias directrices que se detallan en la Tabla 1 y que están siendo actualizadas constantemente. Posteriormente, en el 2009, la Organización Mundial de la Salud (OMS) realiza lo mismo, y en el 2012, la Administración de Medicamentos y Alimentos de los EE.UU. (FDA) publica sus normativas.
En América Latina, en la última década, las autoridades reguladoras comenzaron a definir las reglamentaciones para el registro de un mercado de BS14, 15. En general, los países tienden a adoptar normas internacionales aceptadas. Sin embargo, hay países como Brasil16 y Argentina con procesos regulatorios más consolidados, y otros, como Paraguay y Bolivia, con proyectos en fase de desarrollo17. En la Tabla 2 se muestran comparativamente las legislaciones de EMA, FDA y las de Argentina.
Marco legal en Argentina
En Argentina18, las dos organizaciones responsables de la evaluación de BS son la ANMAT (Administración Nacional de Medicamentos, Alimentos y Tecnología Médica), que es el organismo regulador que da la concesión de licencias de BS, y el Instituto Nacional de Medicamentos, que es responsable de las evaluaciones técnicas de estos productos. En 2011 y 2012, la ANMAT publica las diferentes reglamentaciones, que se detallan en la Tabla 3. En dicha legislación se establece que no es necesario que los BS experimenten el mismo rigor clínico que un medicamento innovador, por lo cual los BS están sujetos a estudios comparativos con la referencia biológica para demostrar la bio-similitud.
Estos estudios comparativos incluyen ensayos preclínicos, así como estudios de fase I, II y III. Dicha legislación establece que para cada posible indicación de un BS se requerirán datos científicos que sustenten cada una de ellas.
Cómo se procedió a la reunión de expertos
El panel de expertos tuvo dos coordinadoras por parte de SOLAPSO que fueron responsables de:
– Seleccionar a los expertos: basándose en su experiencia en el control y tratamiento de psoriasis en placa moderada a grave y en el manejo de biológicos, asegurando la representación de diferentes prácticas clínicas.
– Definición de los temas principales, asignación de los expertos a cada tema y envío de la respectiva literatura: extrapolación de indicaciones, inmunogenicidad, intercambiabilidad, nomenclatura/ trazabilidad.
– Supervisión del Proyecto
El rol de los expertos (nueve en total incluyendo las dos coordinadoras) fue revisar la literatura seleccionada, preparar las respuestas específicas que reflejaran los tópicos más controversiales dentro de cada tema, y proponer una respuesta adecuada para cada pregunta. La cantidad de preguntas para cada tema fueron entre cuatro y siete.
La búsqueda bibliográfica se enfocó en identificar artículos de revisión que abarcaran los cuatro temas elegidos, posicionamientos de diferentes sociedades médicas del mundo, guías sobre biosimilares de la EMA, OMS y FDA, y artículos científicos (ensayos clínicos y estudios observacionales) conducidos en esta área.
Durante la reunión del Consenso de Expertos se realizó una presentación mostrando las preguntas y respuestas preliminares, con el fin de facilitar la discusión. Los expertos debatieron, aportaron su visión y lograron



consenso. Se resaltaron los temas controversiales, que fueron volcados en este manuscrito.
La Consultora Científica Eurotrials brindó soporte metodológico, incluyendo las búsquedas bibliográficas, preparación de la reunión y recolección de datos.
Controversias
Extrapolación de indicaciones
El término extrapolación, según la Real Academia Española, significa aplicar a un ámbito determinado conclusiones obtenidas en otro. En el caso de BS, la extrapolación se refiere a proyectar los datos y resultados recolectados en una indicación clínica aprobada sobre el beneficio de una droga, eficacia y seguridad, a otras indicaciones sin conducir ensayos clínicos adicionales19. Esto es posible cuando las evidencias aportadas son suficientemente persuasivas y deben ser justificadas según los mecanismos de acción, farmacocinética y dinamia, fisiopatología de la enfermedad, perfil de seguridad y experiencia clínica con el fármaco de referencia. La extrapolación reduce el número de ensayos clínicos necesarios y por lo tanto disminuye los costos, como también reduce el impacto financiero de la terapia biológica en los sistemas de salud y, en consecuencia, podría facilitar el acceso a estos fármacos para los tratamientos.
Sin embargo, existen dudas en este punto. Una de ellas es si la similitud bio-analítica coincide con la eficacia y seguridad clínica comprobada. Y otra, es si se puede garantizar la seguridad y la eficacia en otras indicaciones. Los biosimilares disfrutan de la ventaja conceptual de tener un comparador conocido (fármaco original o de referencia) con estructura bien definida, función biológica, seguridad clínica y eficacia. Al no estar disponibles para los fabricantes de biosimilares los lotes originales del producto de referencia, la comparabilidad podría comprometerse20.
Para que la extrapolación de los BS sea considerada válida, el mecanismo de acción, farmacocinética y dinamia (incluida la biodistribución), la inmunogenicidad y la seguridad necesitan ser similares a las del fármaco original, a la indicación extrapolada y a las indicaciones clínicas testeadas. Si uno de estos parámetros es diferente, se requerirían evidencias adicionales para realizar la extrapolación20. La validez del uso de la extrapolación de estudios clínicos de una determinada enfermedad para aprobar un BS en otra enfermedad puede evaluarse a través de las siguientes preguntas:
¿Los mecanismos de acción, farmacocinética, farmacodinamia, inmunogenicidad y seguridad del BS entre la enfermedad que se quiere extrapolar (por ejemplo, psoriasis) y la población clínicamente probada (por ejemplo, artritis reumatoidea) son similares?;
Después de la aprobación, ¿las pruebas de post-comercialización (programas de vigilancia y otros programas de la vida real como los registros) proporcionan información preocupante?20.
Hay situaciones donde no puede asegurarse la extrapolación y se requieren estudios adicionales para definirla21 como, por ejemplo:
Cuando existen distintos mecanismos de acción, en donde la extrapolación debe evaluarse caso por caso.
Si los productos son para especialidades diferentes
Si el mecanismo de acción no es totalmente conocido.
Si existe el riesgo de sub-tratamiento o variados perfiles de seguridad o grupos de pacientes diferentes.
Tanto la EMA como la FDA abogan por la extrapolación y han desarrollado medidas para lograrlo. Para ambas entidades es necesario disponer de al menos un ensayo clínico de equivalencia con la potencia necesaria para determinar la comparabilidad de ambos productos en cuanto a eficacia y seguridad, que deben ser tanto “no inferiores” como “no superiores” al producto de referencia. Además, en dichas guías, es claro que si la bio-similitud se ha demostrado en una indicación, la extrapolación a otras indicaciones del producto de referencia podría ser aceptable con justificación científica apropiada. En cuanto a la inmunogenicidad, se debe evaluar durante el ensayo de seguridad según la EMA, mientras que la FDA solicita un ensayo comparativo previo a la comercialización y otro posterior a la misma, y en ambos casos se exige el desarrollo de un programa de fármaco-vigilancia post comercialización22. En conclusión, los datos en farmacocinética, farmacodinamia, seguridad, eficacia e inmunogenicidad deberían ser obtenidos en la población más sensible para demostrar similitud a la molécula original, y para extrapolar a otras indicaciones22. La extrapolación de datos clínicos de una indicación a otra requiere consideraciones cuidadosas y una sólida justificación científica. Las nuevas indicaciones aprobadas son importantes para el prescriptor a fin de evaluar las opciones terapéuticas, y para tomar precauciones en el futuro. En Argentina, la legislación establece que cada posible indicación de un BS para la cual se solicita autorización, deberá ser la misma para la cual fuera aprobado el producto referente.
Inmunogenicidad
La inmunogenicidad es la capacidad de un compuesto para desencadenar una respuesta inmune, que puede ser de tipo celular y/o humoral. Todos los agentes biológicos generan inmunogenicidad, aun los anticuerpos humanizados23.
Existen múltiples factores que influyen en el desarrollo de la inmunogenicidad. Algunos dependen del paciente, como factores genéticos, edad, exposición previa a fármacos similares, enfermedad y uso de fármacos inmunosupresores concomitantes. Otros dependen del fármaco y de su proceso de fabricación, como por ejemplo la glicosilación y la presencia de agregados y/o contaminantes en la producción del agente biológico, entre otros. Por esta razón, sutiles alteraciones en la manufactura de estas drogas, podrían alterar el patrón de inmunogenicidad24.
Las entidades regulatorias, tanto EMA25 como FDA26, desarrollaron directrices para evaluar la inmunogenicidad. La inmunogenicidad no puede ser predicha por estudios en animales; por lo tanto, se requieren datos de la respuesta inmune en humanos. Si un evento, debido a la respuesta inmune desencadenada, es detectado en la fase pre-aprobación, el mismo puede ser controlado. Por ejemplo, ante el hallazgo de excesiva inmunogenicidad de un BS, se realiza un paso adicional en la purificación, corrigiendo dicho evento. Pero hay casos donde la incidencia es rara, entonces se requerirán estudios de seguridad post-comercialización para detectarla19.
La inmunogenicidad de estos tratamientos se traduce en el desarrollo de anticuerpos contra estos fármacos, anticuerpos antidroga (AA), muy variables en su incidencia, dependiendo de los distintos fármacos y de las técnicas para detectarlos. También son variables las consecuencias que generan, ya que pueden asociarse con aumento o disminución en la eliminación de la droga, neutralización del biológico llevando a disminución en la eficacia y efectos adversos como reacciones de hipersensibilidad – sistémicas o locales – sitio de la infusión o inyección23, 27. Esto depende de que el AA sea neutralizante o no neutralizante. Los AA neutralizantes son aquellos que se unen al sitio activo de la proteína terapéutica e inhiben su función, neutralizándola. Como resultado, la eficacia puede verse disminuida con la consecuente falla terapéutica, o asociarse a un aumento de eventos adversos28, 29. Un ejemplo es el caso de infliximab. En estudios realizados con CT-P13, un BS de infliximab, se observó la presencia de AA en un porcentaje similar al observado con infliximab. En los pacientes con AA se demostró una menor eficacia comparada con la observada en aquellos que no desarrollaban AA, pero similar al grupo de personas tratadas con el producto de referencia que desarrollaron AA. Esta asociación se observó en distintas enfermedades tales como la inflamatoria intestinal, la artritis reumatoidea y la espondilitis anquilosante, en los estudios PLANET-AS30 y PLANET-RA31.
Los anticuerpos no neutralizantes se unen a la proteína terapéutica pero no afectan su actividad intrínseca, ni el sitio receptor; es decir, no tienen efectos biológicos, pero pueden formar inmunocomplejos que pueden, a su vez, modificar la farmacocinética de la proteína administrada (ej. aceleran la eliminación del medicamento)32 o aumentar las reacciones adversas al agente. Tal es el caso de anticuerpos anti-adlimumab, que pueden aumentar el riesgo de eventos tromboembólicos en pacientes con artritis reumatoide y artritis psoriásica33, o la asociación de AA con mayores reacciones durante la infusión de infliximab. Estos efectos se han observado también en estudios comparativos con CT-P13, donde se detectaron mayores reacciones en pacientes con anticuerpos contra BS, pero en porcentajes similares a los encontrados en infliximab31.
En un estudio que evaluó los AA contra remsima (CT-P13), uno de los BS de infliximab, se demostró un patrón de inmunogenicidad semejante al originador34. Sin embargo, en otros casos como el del SB4, el BS de etanercept, la presencia de anticuerpos es inferior al producto original (0.7 vs. 13.1%)35. Las diferencias en inmunogenicidad, tanto en mayor como en menor grado, marcarían la disparidad entre la droga originadora y el BS, que podría alterar la eficacia y seguridad del producto BS. Por este motivo, la comparación de la capacidad de formación de anticuerpos entre estas drogas, es decir la evaluación de inmunogenicidad, es esencial para determinar su aprobación como biosimilares, sobre todo si se plantea la intercambiabilidad entre la droga original y el BS. La magnitud y característica de la respuesta inmune dependerá principalmente de la similitud entre ambas drogas. Pequeñas diferencias en la manufactura podrían desencadenar una respuesta clínica diferente, ya sea con mayor o menor eficacia, o un patrón distinto de efectos adversos. Lo que se busca al intercambiar la droga, es tener una respuesta predecible y similar, por lo cual la intercambiabilidad requiere estudios de inmunogenicidad cruzada, de corto y largo plazo. En teoría, cualquier estudio de inmunoensayo puede ser utilizado para medir anticuerpos anti droga. Sin embargo, los estudios que detectan anticuerpos contra anticuerpos monoclonales pueden ser técnicamente problemáticos, ya que muchas veces el anticuerpo para detectar el anti- inmunoglobulinas se une al producto mismo, dificultando su detección, por lo que las pruebas de ELISA o radioinmunoprecipitación no son útiles, a menos que se hayan corregido para medir anticuerpos monoclonales. Una técnica común, es la de utilizar la prueba de bridging, que no requiere reactivos anti-inmunoglobulinas y se aplica en forma directa para detectar los anticuerpos. El problema es que este formato puede ser menos sensible y requiere un gran esfuerzo técnico para ser desarrollado36. En otras ocasiones, las muestras de plasma o suero, pueden contener fragmentos proteicos que generen falsos resultados positivos o negativos, por lo que estos estudios requieren ser hechos a medida, para detecciones específicas25.
En el caso del infliximab30, 31, los estudios de extensión PLANET-AS y PLANET-RA, han evaluado este ítem. La intercambiabilidad no derivó en cambios clínicos significativos, y la adición de metotrexato, conocido por disminuir la formación de anticuerpos antidroga, tuvo efectos similares en infliximab y CT-P13, su biosimilar. En un estudio de enfermedad inflamatoria intestinal, se observó la presencia de anticuerpos antidroga al intercambiar infliximab por CT-P13. Por este motivo es fundamental establecer un programa de farmacovigilancia que pueda detectar diferencias en el curso de tratamientos prolongados7.
Intercambiabilidad
Intercambiabilidad implica que dos productos pueden ser usados en cualquier orden en un paciente, sin considerar la historia del tratamiento.
Por lo tanto, un producto biológico se considera intercambiable con su producto de referencia si36-40 :
a) ese producto biológico es biosimilar al producto de referencia. Esta es una condición previa a la intercambiabilidad, y es necesaria para su aprobación reglamentaria.
b) si se puede esperar que el BS produzca el mismo resultado clínico que el producto original en cada uno de los pacientes.
c) y si mantiene la misma eficacia, seguridad y calidad que su producto de referencia.
El switching es la rotación reiterada entre productos biológicos de referencia y sus BS así como entre BS de un mismo producto de referencia. Esta práctica es desaconsejable, ya que hace imposible la farmacovigilancia, pero ocurre con frecuencia. Las normas de la EMA no hacen referencia a la intercambiabilidad, y dejan las correspondientes decisiones a las autoridades sanitarias de cada país miembro de la Unión Europea. La legislación de EE.UU. permite a la FDA determinar qué biosimilares son intercambiables sin consultar con el médico, pero aún no se ha publicado la normativa definitiva. La intercambiabilidad de los biosimilares dificultará en cualquier caso la farmacovigilancia, por lo tanto, para demostrar la intercambiabilidad de los productos biológicos y para proveer herramientas de asesoramiento regulatorio, se necesitarán más estudios a largo plazo. Si dos productos biológicos ya han sido aprobados y comercializados, las agencias regulatorias de cada país evaluarán qué solicitud adicional presentar que demuestre que se cumplen las condiciones para su intercambiabilidad36-38. Parece prudente dejar al médico tratante especialista, como el único prescriptor de la eventual intercambiabilidad, avalar su decisión por accesibilidad al producto biológico o biosimilar. Se desaconseja formalmente la posibilidad de que sea indicada por obras sociales, sistemas prepagos, farmacéuticos, centros expendedores y auditores médicos, considerando la complejidad de la indicación y farmacovigilancia a futuro37, 38. El paciente debe ser informado de la decisión y necesidad del intercambio.
Nomenclatura y trazabilidad
La trazabilidad posibilita seguir el rastro de un medicamento desde la fabricación a través de todas las etapas de distribución hasta su consumo, contribuyendo a mejorar la eficiencia y la seguridad del paciente en el proceso terapéutico. Las sustancias activas de los medicamentos biológicos son más grandes y más complejas que las de los medicamentos no biológicos, así como el proceso de fabricación. Ello puede dar lugar a que pequeñas diferencias entre las moléculas de la misma sustancia activa, puedan repercutir en el perfil de seguridad. En ello se fundamenta la importancia de una adecuada identificación del producto y su trazabilidad.
Sin embargo, en los BS, la nomenclatura ha sido objeto de controversia41, 42. Habitualmente, a las sustancias biológicas se les asignan una Denominación Común Internacional, también llamada DCI (o INN, del inglés International Nonproprietary Name). Además, para distinguir entre los perfiles de glicoforma conocidos o susceptibles de ser distintos, se introdujo una segunda palabra griega como parte de la DCI para que diferentes versiones de una glicoproteína tuvieran diferentes DCI. Con la aparición de biosimilares se añadió una complejidad adicional en la nomenclatura. En estos casos, se conceden licencias sobre la base de un estudio de comparabilidad entre el equivalente biosimilar y el producto licenciado. Algunas autoridades utilizan la DCI de la sustancia de referencia sola, mientras que otras consideran que debe darse un identificador distintivo a cada biosimilar. Esto se ha logrado mediante la adición de un calificador que suele ser corto y separado (por ejemplo, en Australia y Japón) y que, en algunos casos incorpora o alude al nombre de la empresa (por ejemplo, en Japón y EE.UU.). Así, en la actualidad, la misma medicina biológica puede tener diferentes identificadores en diferentes partes del mundo.
Para evitar la proliferación de sistemas de calificaciones, en el 2013 algunas autoridades de reglamentación farmacéutica han solicitado al Programa DCI que desarrolle y administre un esquema voluntario y global de nomenclatura complementaria aplicable a los biosimilares para evitar la proliferación de diferentes nombres y políticas de nomenclatura. Este esquema voluntario tiene por objeto proporcionar un código único de identificación llamado calificador biológico (o en inglés, Biological Qualifier), distinto de la DCI, para todas las sustancias biológicas. El calificador biológico (CB) se utiliza como un elemento adicional e independiente utilizado junto con la DCI para una sustancia biológica y solo identifica la sustancia activa en un producto biológico. Dicho código debe consistir en cuatro consonantes aleatorias, es único y permite su uso para la sustancia fabricada en todos los lugares de fabricación que han demostrado que sea comparable, y por todos los titulares de autorizaciones de fabricación que distribuyen productos que contienen la sustancia. Por lo tanto, con el uso del CB, se contribuirá a la identificación de sustancias biológicas para la prescripción y dispensación de medicamentos como para el registro de farmacovigilancia. Este esquema es una decisión voluntaria de cada autoridad regulatoria. A pesar de estos cambios, aún se necesita mayor desarrollo de una política de nomenclatura para facilitar la identificación precisa de todos los productos biológicos y su farmacovigilancia.
Posición de SOLAPSO
SOLAPSO expresa que:
Es importante la aparición de biosimilares en el mercado, para disminuir costos y facilitar el acceso a los pacientes a terapéuticas de alto costo.
Deben cumplirse todas las condiciones para demostrar la bio-similitud con el fármaco de referencia en cuanto a calidad, actividad bilógica, seguridad y eficacia.
La extrapolación no debe darse en forma automática. Debe ser considerada para cada enfermedad.
Mientras no exista información clara acerca del impacto de intercambiar medicamentos biológicos, se desaconseja hacerlo. De no tener otra opción la decisión de realizar el intercambio debe ser consensuada entre el médico tratante, el paciente y el sistema de salud.
Una nomenclatura diferenciada es fundamental para una correcta farmacovigilancia.
Agradecimientos: Este trabajo fue financiado por el Laboratorio Abbvie.
Se agradece a Eurotrials, Scientific Consultants su colaboración en la planificación y orientación metodológica del proyecto.
Conflicto de intereses: La Dra. Nélida Raimondo he actuado como conferencista o brindando asesoramiento en temas relacionados al actual trabajo para Pfizer y Abbvie. Los Dres. Cristina Echeverría y Luis Mazzuoccolo son conferencistas de Abbvie y Novartis. El Dr. Matías Maskin participa en Advisory boards de Novartis y Eli Lilly. Tiene honorarios como conferencista de Abbvie, Novartis, La Roche Possay y Vichy. La Dra. Nora Kogan es investigadora principal y conferencista de Abbvie, Eli Lilly, Janssen, Novartis y Pfizer. La Dra. Paula Luna es oradora de Abbvie y Novartis, e investigadora en Trials de Novartis y Eli Lilly.
Bibliografía
1. Pasini L, Casadei G, Nobili A. Biological agents and biosimilars: essential information for the internist. Eur J Intern Med 2016; 33: 28-35.
2. Mellstedt H. Clinical considerations for biosimilar antibodies. EJC Suppl 2013; 11: 1-11.
3. Isaacs JD, Cutuolo M, Keystone EC, Park W, Braun J. Biosimilars in immune-mediated inflammatory diseases: initial lessons from the first approved biosimilar anti-tumour necrosis factor monoclonal antibody. J Intern Med 2016; 279: 41-59.
4. Huggett B. Public biotech 2012 – the numbers. Nat Biotechnol 2013; 31: 697-703.
5. Committee for Medicinal Products for Human Use (CHMP). Guideline on similar biological medicinal products [Internet]. London, UK: European Medicines Agency (EMA); 2014 Oct [cited 2017 May 29] p. 7. Report No.: CHMP/437/04 Rev 1.En: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf; consultado enero 2018.
6. Committee for Medicinal Products for Human Use (CHMP). Assessment report – Inflectra. International non-proprietary name: Infliximab [Internet]. London: European Medicines Agency (EMA); 2013 Jun [cited 2017 May 17] p. 105. Report No.: EMA/CHMP/589422/2013. En: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002778/WC500151490.pdf; consultado enero 2018.
7. Babini A, Kos IA, Matar P, Teixeira FV, Feijó Azevedo V. New monoclonal antibody biosimilars approved in 2015 in Latin America: position statement of the Latin American Forum on Biosimilars on biosimilarity, interchangeability and extrapolation of indications. GaBI Journal 2016; 5: 66-9.
8. European Medicines Agency (EMA). European public assessment reports [Internet]. 2018 [cited 2018 Jun 5]. En: http://www.ema.europa.eu/ema/index.jsp?searchType=name&taxonomyPath=&genericsKeywordSearch=Submit&searchGenericType=biosimilars&keyword=Enter+keywords&alreadyLoaded=true&curl=pages%2Fmedicines%2Flanding%2Fepar_search.jsp&status=Authorised&mid=WC0b01ac058001d124&treeNumber=&searchTab=searchByAuthType&pageNo=2; consultado febrero 2018.
9. Gunn G 3rd, Sealey DC, Jamali F, Meibohm B, Ghosh S, Shankar G. From the bench to clinical practice: understanding the challenges and uncertainties in immunogenicity testing for biopharmaceuticals. Clin Exp Immunol 2016; 184: 137-46.
10. Kurki P, Ekman N. Biosimilar regulation in the EU. Expert Rev Clin Pharmacol 2015; 8: 649-59.
11. Alten R, Cronstein BN. Clinical trial development for biosimilars. Semin Arthritis Rheum 2015; 44: S2-8.
12 Declerck P, Mellstedt H, Danese S. Biosimilars – terms of use. Curr Med Res Opin 2015; 31: 2325-30.
13. Rak Tkaczuk KH, Jacobs IA. Biosimilars in oncology: from development to clinical practice. Semin Oncol 2014; 41(Suppl 3): S3-12.
14. Aceituno Ávarez A, Mysler E, Ruiz de Castilla EM, Flores-Murrieta FJ, Hughes J, Feijó Azevedo V. Recommendations for the regulation of biosimilars and their implementation in Latin America. GaBI Journal 2014; 3: 143-8.
15. Bennett CL, Chen B, Hermanson T, et al. Regulatory and clinical considerations for biosimilar oncology drugs. Lancet Oncol 2014; 15: e594-605.
16. Araujo DV. Consideraciones regulatorias sobre productos biológicos en Brasil. Value Health Reg Issues 2012; 1: 254-6.
17. Garcia R, Araujo DV. The Regulation of biosimilars in Latin America. Curr Rheumatol Rep 2016; 18: 16.
18. Feijó Azevedo V, Sandorff E, Siemak B, Halbert RJ. Potential regulatory and commercial environment for biosimilars in Latin America. Value Health Reg Issues 2012; 1: 228-34.
19. González Andrade F. Consideraciones clínicas en la terapia con biológicos y biosimilares. En: Medicamentos biológicos: presente y futuro de la terapéutica. 1st ed. Quito, Equador: Litocolor, 2017, p 319.
20. Ben-Horin S, Vande Casteele N, Schreiber S, Lakatos PL. Biosimilars in inflammatory bowel disease: facts and fears of extrapolation. Clin Gastroenterol Hepatol 2016; 14: 1685-96.
21. Dörner T, Strand V, Castañeda-Hernández G, et al. The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis 2013; 72: 322-8.
22. Reinish W, Louis E, Danese S. The scientific and regulatory rationale for indication extrapolation: a case study based on the infliximab biosimilar CT-P13. Expert Rev Gastroenterol Hepatol 2015; 9 (Suppl 1): 17-26.
23. Ryff JC, Schellekens H. Immunogenicity of rDNA-derived pharmaceuticals. Trends Pharmacol Sci 2002; 23: 254-6.
24. Jani M, Barton A, Warren RB, Griffiths CW, Chinoy H. The role of DMARDs in reducing the immunogenicity of TNF inhibitors in chronic inflammatory diseases. Rheumatology (Oxford) 2014; 53: 213-22.
25 Committee for Medicinal Products for Human Use (CH. Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical [Internet]. London, UK: European Medicines Agency (EMA); 2012 May [cited 2017 May 17] p. 10. Report No.: EMA/CHMP/BMWP/86289/210. En: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128688.pdf; consultado febrero 2018.
26. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product [Internet]. Food and Drug Administration; 2015 Apr [cited 2017 May 17] p. 27. En: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf; consultado febrero 2018.
27. Porter S. Human immune response to recombinant human proteins. J Pharm Sci 2001; 90: 1-11.
28. Vincent FB, Morand EF, Murphy K, Mackay F, Mariette X, Marcelli C. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)-specific neutralising agents in chronic inflammatory diseases: a real issue, a clinical perspective. Ann Rheum Dis 2013; 72: 165-78.
29. Garcês S, Demengeot J, Benito-Garcia E. The immunogenicity of anti-TNF therapy in immune-mediated inflammatory diseases: a systematic review of the literature with a meta-analysis. Ann Rheum Dis 2013; 72: 1947-55.
30. Park W, Hrycaj P, Kovalenko V, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis 2013; 72: 1605-12.
31. Yoo D, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis 2013; 72: 1613-20.
32. Schellekens H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clin Ther 2002; 24: 1720-40.
33. Korswagen LA, Bartelds GM, Krieckaert CL, et al. Venous and arterial thromboembolic events in adalimumab-treated patients with antiadalimumab antibodies: a case series and cohort study. Arthritis Rheum 2011; 63: 877-83.
34. Ben-Horin S, Yavzori M, Benhar I, et al. Cross-immunogenicity: antibodies to infliximab in Remicade-treated patients with IBD similarly recognise the biosimilar Remsima. Gut 2016; 65: 1132-8.
35. Emery P, Vencovsk. J, Sylwestrzak A, et al. A phase III randomised, double-blind, parallel-group study comparing SB4 with etanercept reference product in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis 2017; 76: 51-7.
36. Puig L. Biosimilars and reference biologics: decisions on biosimilar interchangeability require the involvement of dermatologists. Actas Dermosifiliogr 2014; 105: 435-7.
37. Chow SC. Assessing biosimilarity and interchangeability of biosimilar products. Stat Med 2013; 32: 361-3.
38. Tothfalusi L, Endrényi L, Chow SC. Statistical and regulatory considerations in assessments of interchangeability of biological drug products. Eur J Health Econ 2014; 15 Suppl 1: S5-11.
39. Braun J, Kudrin A. Switching to biosimilar infliximab (CT-P13): evidence of clinical safety, effectiveness and impact on public health. Biologicals 2016; 44: 257-66.
40. Benucci M, Gobbi FL, Bandinelli F, et al. Safety, efficacy and immunogenicity of switching from innovator to biosimilar infliximab in patients with spondyloarthritis: a 6-month real-life observational study. Immunol Res 2017; 65: 419-22.
41 Casadevall N, Felix T, Strober BE, Warnock DG. Similar names for similar biologics. BioDrugs 2014; 28: 439-44.
42. Technologies Standards and Norms (TSN), Regulation of Medicines and other Health Technologies (RHT), Essential Medicines and Health Products (EMP), World Health Organization (WHO). Biological qualifier: an INN proposal – programme on Inernational Nonproprietary Names (INN) [Internet]. Geneva: WHO; 2015 Oct [cited 2017 May 17] p. 5. Report No.: 14.342. En: http://www.who.int/medicines/services/inn/WHO_INN_BQ_proposal_2015.pdf; consultado febrero 2018.
43. Committee for Medicinal Products for Human Use (CHMP). Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1) [Internet]. London, UK: European Medicine Agency (EMA); 2014 May [cited 2017 May 29] p. 9. Report No.: EMA/CHMP/BWP/247713/2012. En: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/06/WC500167838.pdf; consultado febrero 2018.
44. Committee for Medicinal Products for Human Use (CH. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues [Internet]. London, UK: European Medicines Agency (EMA); 2014 Dec [cited 2017 May 17] p. 13. Report No.: EMEA/CHMP/BMWP/42832/2005 Rev1. En: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf; consultado febrero 2018.
45. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for industry: clinical pharmacology data to support a demonstration of biosimilarity to a reference product [Internet]. Food and Drug Administration; 2014 May [cited 2017 May 17] p. 18. Report No.: Draft guidance. En: http://patentdocs.typepad.com/files/ucm397017.pdf; consultado febrero 2018.
46. Committee for Medicinal Products for Human Use (CH. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues [Internet]. London: European Medicines Agency (EMA); 2012 May [cited 2017 May 17] p. 16. Report No.: EMA/CHMP/BMWP/403543/2010. En: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf; consultado febrero 2018.
47. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for industry: biosimilars: questions and answers regarding implementation of the biologics price competition and innovation act of 2009 [Internet]. Food and Drug Administration; 2015 Apr [cited 2017 May 17]. En: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM444661.pdf; consultado febrero 2018.
48. Committee for Medicinal Products for Human Use (CH. Concept paper on the revision of the guideline on similar biological medicinal product [Internet]. London, UK: European Medicines Agency (EMA); 2011 Nov [cited 2017 May 17] p. 4. Report No.: EMA/CHMP/BMWP/572643/2011. En: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/11/WC500117987.pdf; consultado febrero 2018.
49. European Medicines Agency. Guideline on good pharmacovigilance practices (GVP) – product- or population-specific considerations II: biological medicinal products [Internet]. London: European Medicines Agency (EMA); 2015 Dec [cited 2017 May 17] p. 20. Report No. EMA/168402/2014 DRAFT for public consultation. En: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/12/WC500198757.pdf; consultado febrero 2018.
50. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for industry: nonproprietary naming of biological products [Internet]. Food and Drug Administration; 2015 Aug [cited 2017 May 17] p. 14. En: https://www.pbwt.com/content/uploads/2016/09/10578-drf-08-26-15.pdf; consultado febrero 2018.
– – – –
Que para conocer el país sea necesario estudiarlo, le parecerá al lector una banalidad. Sin embargo, observando lo que ocurre todos los días debe convenir en que la mayoría de sus conciudadanos piensan exactamente lo contrario. Y si se les agrega que es preciso remontarse a los orígenes, seguir paso a paso la evolución interna, para opinar de una manera consciente sobre el fenómeno contemporáneo, no es imposible que una discreta sonrisa sea la única respuesta. Excuso decir al lector que en esto, como en lo demás, el libro de la vida permanece cerrado para el que no se afana en descifrarlo.
Juan Agustín García (1862-1923)
La ciudad indiana (1900). Buenos Aires: Emecé, s/f, p 11