JULIÁN MAGGIO, NAZARENO GONZÁLEZ, GEORGINA A. CARDAMA, DANIEL E. GOMEZ
Laboratorio de Oncología Molecular, Departamento de Ciencia y Tecnología,
Universidad Nacional de Quilmes, Buenos Aires, Argentina
Resumen Las Rho GTPasas son una familia de proteínas que actúan como interruptores moleculares en
diversas vías de señalización coordinando la regulación de distintos procesos celulares. La desregulación de dichas proteínas se vincula con transformación maligna y progresión tumoral en distintos tipos de cáncer. Por estos motivos, en los últimos años las Rho GTPasas fueron postuladas como blancos moleculares interesantes. En este trabajo describimos las distintas estrategias estudiadas utilizando a las Rho GTPasas como blanco y su grado de avance, mostrando una estrategia novedosa para el tratamiento del cáncer.
Palabras clave: Rho GTPasas, cáncer, blanco molecular
Abstract Rho GTPases as molecular targets in cancer. Strategies and therapeutic opportunities. Rho
GTPases are molecular switches that control the different cellular processes. Deregulation of these proteins is associated to transformation and malignant progression in several cancer types. Given the evidence available of the role of Rho GTPases in cancer it is suggested that these proteins can serve as potential therapeutic targets. This review focuses on the strategies used to develop Rho GTPases modulators and their potential use in therapeutic settings.
Key words: Rho GTPases, cancer, molecular target
Recibido: 5-VII-2017 Aceptado: 23-X-2017
Dirección postal: Dr. Daniel Gomez, Laboratorio de Oncología Molecular, Universidad Nacional de Quilmes, R. Sáenz Peña 352, 1876 Bernal, Buenos Aires, Argentina
e-mail: degomez@unq.edu.ar
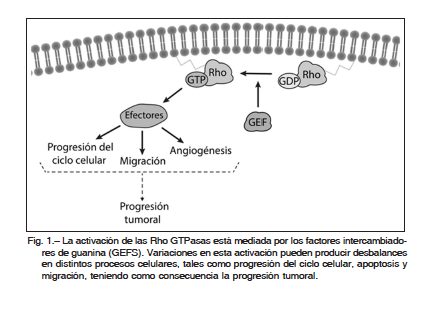
Las pequeñas GTPasas de la superfamilia Ras son proteínas de 21 kDa que funcionan como interruptores moleculares en un gran número de vías de señalización. Dentro de esta superfamilia se encuentran las proteínas Rho (Ras homologous)1. Las Rho GTPasas son interruptores moleculares que adoptan diferentes estados conformacionales en respuesta a la unión de GDP o GTP. Solo en el estado activo unido a GTP, estas proteínas son capaces de unirse y activar a sus proteínas efectoras. Es importante destacar que muchas de las funciones de las Rho GTPasas requieren del anclaje a membrana para ser funcionales. La actividad de las Rho GTPasas está estrictamente regulada a fin de estimular local y temporalmente a diferentes efectores celulares. Hasta el momento se conocen tres clases de proteínas que regulan a las Rho GTPasas: factores intercambiadores de guanina (GEFs o Guanosine Exchange Factors), proteínas activadoras de GTPasas (GAPs o GTPase activating proteins) e inhibidores de disociación de guanina (GDIs o Guanine dissociation inhibitors).
Cuando las proteínas Rho se encuentran activas y ancladas a la membrana, son capaces de interactuar con diferentes efectores río abajo. Se conocen más de 70 efectores potenciales para las proteínas de la familia Rac1 y Rho. En este sentido, cada Rho GTPasa es capaz de activar un grupo definido de efectores y estos efectores pueden ser tanto efectores catalíticos (ej. quinasas como Pak1, activada por Rac1; la quinasa ROCK, activada por RhoA, entre otras) como efectores no catalíticos que funcionan como adaptadores o scaffolds (ej., Diaphanous, Was, Baiap2, entre otros)2.
Tres miembros de la familia de Rho GTPasas han sido estudiados en detalle: RhoA, Cdc42 y Rac1. Tradicionalmente, Rac1 ha sido descripta como uno de los reguladores principales de la reorganización del citoesqueleto de actina, específicamente en la formación de lamelipodios que contribuyen de manera pivotal a la migración celular. En este sentido, un ligando extracelular como el factor de crecimiento epidérmico (EGF) o insulina, provocan la formación de lamelipodios y ondulaciones de membrana a través de Rac13. De este rol importante en la regulación del citoesqueleto se desprende que Rac1 está involucrada en la regulación de procesos como endocitosis, tráfico de membrana, morfología celular, adhesión, spreading y polaridad celular, destacándose la regulación de la migración celular4-6.
Rho GTPasas y cáncer
Las Rho GTPasas tienen un rol central en la regulación de diversos procesos celulares y su alteración o desbalance puede ser la causa molecular de diversas enfermedades, incluyendo al cáncer (Fig. 1). En este sentido, las Rho GTPasas controlan algunos de los procesos fundamentales de la célula a través de una serie de complejos mecanismos bioquímicos. Las mutaciones o alteraciones de este complejo sistema de señalización podrían conducir al desarrollo de cáncer. Tradicionalmente, la sobreactivación de la vía de las Rho GTPasas ha sido asociada a la transformación maligna. Recientemente, se ha descrito una mutación con ganancia de función en el gen rac1 en pacientes con melanoma provocado por exposición a radiación solar. Sin embargo, las mutaciones de rac1 se encuentran con poca frecuencia en otros tipos de cánceres humanos7-9. La regulación positiva de Rac1 se debe mayormente a alteraciones de sus proteínas reguladoras. Un ejemplo representativo de esto es la sobreactivación de sus activadores tipo GEF. Esta activación es comúnmente dirigida por señales anómalas de los receptores de factores de crecimiento (RTKs) y receptores acoplados a proteína G (GPCRs), o bien por la mutación de los GEFs1. En este aspecto, muchos GEFs presentan un rol relevante en cáncer, como Ect2, Tiam1, la familia de proteínas Vav, P-Rex1, la familia de proteínas DOCK, entre otros10-13.
Las primeras evidencias de transformación mediada por Rho GTPasas fueron provistas por experimentos en los que Rac1 constitutivamente activo, y en menor medida RhoA, indujeron transformación maligna en fibroblastos y tumorigenicidad en ratones atímicos1,9. Siguiendo con esta idea, Rac1 está implicado en la transición epitelio-mesenquimal y en la transición mesenquimal-epitelial14-17.
Actualmente, las alteraciones en la vía de Rho GTPasas pueden ser encontradas en muchos tipos de cáncer. Entre ellos se puede mencionar tumores malignos de páncreas, mama, piel, próstata, testículo, colon, hígado, pulmón, estómago, así como también en leucemias, osteosarcoma, neuroblastoma, glioblastoma y cáncer de cabeza y cuello18-21. A pesar de que la sobreexpresión de las Rho GTPasas se observa comúnmente en el cáncer, la regulación negativa de algunos de los miembros de la familia de Rho también ha sido asociada a cáncer22.
La pérdida de la polaridad celular es uno de los sellos distintivos del cáncer, un fenómeno común en las células de carcinoma23, 24. Para que las células mantengan su polaridad debem mantener su contacto célula-célula; siendo Rac1, RhoA y Cdc42 indispensables para conservar un estado normal25.
Otro de los puntos importantes de la progresión tumoral es la migración e invasión. En este sentido, la vía de RhoA/ROCK tiene un papel central en la adquisición de propiedades migratorias e invasivas en células tumorales26, ya que están implicadas en la remodelación del citoesqueleto de actina, en la adhesión intracelular mediada por cadherinas y en la remodelación de la matriz extracelular (ECM), especialmente por regulación de la liberación de metaloproteasas de la matriz 9 (MMP-9)27. En este sentido, se ha informado que Rac1 es una proteína clave en el proceso invasivo de meduloblastoma infantil28. En modelos de gliobastoma multiforme Rac1, Rac3 y algunos GEFs regulan la capacidad migratoria e invasiva de este tipo de tumores12, 29, 30.
Una vez que se establece un tumor, la neovascularización del tejido patológico es esencial31. Se ha identificado un rol importante de las proteínas Rho en
la formación de capilares dependientes del factor de crecimiento endotelial vascular (VEGF) en estudios in vitro e in vivo32.
En cuanto a las proteínas regulatorias de las GTPasas involucradas en cáncer, uno de los ejemplos más estudiados es el de Rac1-GEF Tiam1 (invasión y metástasis de linfoma de células T). Aunque las GTPasas no son consideradas oncogenes, tiam1 ha sido confirmado como uno, ya que se encuentra implicado en la transformación oncogénica de fibroblastos33, 34. Se estableció que Tiam1 incrementa la capacidad invasiva y el potencial metastásico de las células tumorales35. Adicionalmente, los niveles de Tiam1 se encuentran aumentados y correlacionan con el pronóstico de carcinomas humanos de próstata, carcinomas nasofaríngeos y con el grado de progresión de tumores de mama36. Siguiendo con esta idea, Vav1, otro GEF de Rac1, se encuentra sobreexpresado en adenocarcinomas pancreáticos y en melanomas metastásicos37.
También se ha informado un rol de las proteínas GAP en la progresión tumoral. Este es el caso de β2-quimerina, un Rac1-GAP, que se encuentra significativamente disminuida en tumores mamarios38. Adicionalmente, se ha demostrado que la sobreexpresión de β2-quimerina en un modelo murino de carcinoma mamario produce una reducción de la tasa de crecimiento tumoral y de su capacidad invasiva y metastásica39.
Otra característica distintiva exhibida por las células tumorales es el potencial replicativo ilimitado debido principalmente a la actividad de la enzima telomerasa. Esta enzima mantiene el largo del telómero, agregando repeticiones TTAGGG al final de los cromosomas en cada división celular40. Además de esta función, existen roles extrateloméricos de la telomerasa que están involucrados en la promoción del cáncer41. Se ha demostrado que la expresión de la telomerasa aumenta la migración de fibroblastos a través de la activación del eje CXCL12/CXCR4 y la familia de las proteínas Rho42.
Yeh y col. demostraron que Cdc42 y Rac1 participan en el control de la actividad de la telomerasa en células tumorales nasofaríngeas humanas (NPC-076). Este grupo investigó los efectos de la inhibición de Cdc42 y Rac1 sobre la actividad de la telomerasa en las células. Inhibiendo la expresión de Cdc42 o Rac1 redujo la actividad de la telomerasa en las células NPC-07643. Esta inhibición no fue asociada con una disminución de la transcripción del gen hTERT (transcriptasa reversa de la telomerasa), el componente clave de la telomerasa44. Esto sugiere que Cdc42/Rac1 participan en el control post-transcripcional de la actividad de la telomerasa en NPC-076. Por otro lado, también se propone que la sobreexpresión de Rac3 es requerida para el mantenimiento de la actividad de la telomerasa45. Análisis adicionales sugieren que la expresión exógena de hTERT puede promover la capacidad de invasión y metástasis a través de un aumento en la regulación de MMP9 y RhoC46.
Dado que la telomerasa es un blanco clave en cáncer, se han desarrollado varias moléculas dirigidas al mismo. Recientemente, se ha postulado que la regulación de la telomerasa puede ser mejor que la inhibición directa47. Por el momento no se ha determinado si las Rho GTPasas pueden ejercer esta acción de una forma más efectiva que las conocidas hasta ahora.
Entre todos los efectores de Rho GTPasas, cuatro de ellos están directamente relacionados con la transformación maligna. En primer lugar se puede mencionar la familia WASP, efectores de Rac1 y Cdc42, relacionada con la reorganización del citoesqueleto de actina, por lo tanto asociada a la migración celular y formación de filopodios y podosomas. Otro efector descripto es IQGAP1, el cual está implicado en rearreglos de membrana por su interacción con Rac1 y Cdc42 y en las uniones célula-célula mediadas por E-cadherinas48, 49.
En tercer lugar, se debe mencionar el papel de Pak, que en cáncer ha sido extensamente estudiado. Estas quinasas tienen más de 40 posibles sustratos y regulan procesos biológicos como la proliferación celular, la motilidad celular, la supervivencia, angiogénesis y crecimiento independiente de sustrato, entre otros50. Se ha descripto que mediante la activación de Rac1, Pak1 es capaz de activar a la proteína quinasa Cγ (PKCγ), un evento necesario para la interacción con la proteína fascina, mediando la migración celular en un modelo humano de carcinoma de colon51.
Finalmente, ROCK1 interactúa directamente y estabiliza el oncogen c-Myc conduciendo a un aumento de la expresión miR-17-92 en cáncer de mama y próstata52; los miembros de la familia ROCK también son efectores directos de RhoA53.
Es importante remarcar que Rac1 puede también conducir a efectos antitumorales54. Recientemente Zandvakiliy col. analizaron el papel de Rho GTPasas con potenciales funciones de supresión tumoral55. De todas maneras la función redundante entre Rho GTPasas, sus múltiples reguladores y efectores sumados a las vías de señalización vinculadas a esta familia de proteínas pueden explicar dichas contradicciones.
Rho GTPasas como blancos terapéuticos en cáncer
Las evidencias que relacionan a las Rho GTPasas con cáncer las han convertido en un atractivo blanco terapéutico. Diversos grupos comenzaron la búsqueda de inhibidores que pudieran influir negativamente en las características del crecimiento de las células tumorales. Algunas estrategias usadas para desarrollar inhibidores o moduladores se describen a continuación.
Inhibición de la interacción Rho GTPasa-GEF
Bloquear la interacción de una Rho GTPasa con su GEF específico representa una vía terapéutica de gran interés. El primer inhibidor caracterizado fue NSC23766.El mismo fue identificado mediante screening virtual56. Se define como screening virtual a la selección de compuestos de grandes bases de datos utilizando herramientas computacionales para predecir su capacidad de unión a un blanco macromolecular57. Esta selección se lleva a cabo utilizando algoritmos que puntúan y clasifican moléculas de grandes bibliotecas de acuerdo a su probabilidad de presentar afinidad por cierto blanco58 .
NSC23766 actúa inhibiendo la activación de Rac1 mediante bloqueo de la asociación de Rac1 y Tiam1, como así también la de Rac1 con Trio, otro GEF de Rac1. NSC23766 ha demostrado tener la capacidad de inhibir efectos pro tumorales en un gran número de modelos de cáncer54. NSC23766 suprimió la migración celular y el crecimiento de cáncer de pulmón de células no pequeñas (NSCLC)59. A pesar de los resultados prometedores asociados con NSC23766, este compuesto carece de la eficacia requerida para su aplicación en la clínica. La optimización de NSC23766 condujo a la identificación de un potente inhibidor de Rac1, EHop-016, que ha demostrado que suprime la migración mediada por Rac1 en células tumorales60. Este inhibidor funciona interfiriendo la unión Rac1-Vav2. Vav2 es un activador tipo GEF de Rac1 que cumple un rol clave en tumores primarios de mama así como también en la formación de metástasis en pulmón61.
También por screening virtual, nuestro grupo ha identificado un inhibidor denominado ZINC69391, capaz de interferir en la unión Rac1-GEF. La validación in vitro demostró que el inhibidor interfiere con la unión Rac1-Tiam162. La inhibición de Rac1 por ZINC69391 mostró una inhibición eficiente de la progresión del ciclo celular, la proliferación y migración celular en líneas celulares de cáncer de mama agresivas. Más aún, el uso de ZINC69391 in vivo redujo significativamente la formación de nódulos pulmonares en modelos murinos de cáncer de mama. Adicionalmente, el uso de ZINC69391 como droga parental nos permitió identificar un análogo más potente, 1A-116, el cual también demostró tener propiedades antimetastásicas in vitro e in vivo. Análisis más exhaustivos de 1A-116 indicaron que el mecanismo de acción consiste en la interferencia de las uniones Rac1-P-Rex1 y la disminución de la activación de Rac163.
Uno de los signos característicos de la progresión tumoral en cáncer de mama y próstata es la transición a la independencia hormonal. Esta transición conlleva a la resistencia a tratamientos anti hormonales. En base a esto, se estudiaron los efectos de 1A-116 en células de cáncer de mama resistentes a tamoxifeno, uno de los agentes terapéuticos más utilizados en el tratamiento del cáncer mamario. La alteración de las vías de señalización de supervivencia cumple un rol fundamental en la resistencia al tamoxifeno, siendo el aumento en la señalización de Rac1-Pak1 una de las alteraciones más importantes. Hemos desarrollado un modelo celular de cáncer de mama (MCF7: C1199) con la actividad de Rac1 potenciada. Estas células mostraron un incremento en los procesos de migración y proliferación celular. Adicionalmente también demostraron que el aumento de la actividad de Rac1 desencadena la transición a un fenotipo hormono-independiente y resistente al tamoxifeno. También hemos demostrado que la actividad de Pak1 se incrementa en respuesta al tamoxifeno, aumentando los niveles de fosforilación de los receptores de estrógeno en la Ser305, un sitio de fosforilación clave en la resistencia al tamoxifeno. 1A-116 restableció de forma efectiva los efectos anti-proliferativos del tamoxifeno, reduciendo la cantidad de Pak1 activo y disminuyendo los niveles de receptores fosfo-Ser305 de estrógeno63. En conclusión, estos datos respaldan el uso de terapias anti hormonales combinadas con inhibidores de Rac1 en los tumores mencionados.También hemos analizado los efectos de ambas: la molécula parental (ZINC69391) y la derivada (1A-116) en gliomas malignos, remarcando la importancia de las señales aportadas por Rac1 en el fenotipo invasivo. Hemos informado que ZINC69391 es capaz de interferir en la interacción de Rac1 con Dock180, un activador importante de Rac1 vinculado a la invasión en glioma, y de reducir los niveles de Rac1-GTP en líneas celulares de glioma humanos. Los niveles de Pak1 también disminuyeron con el tratamiento de ZINC69391. ZINC69391 redujo la proliferación y afectó e inhibió significativamente la invasión y migración celular in vitro, interfiriendo con el citoesqueleto de actina, reteniendo las células en fase G1 y desencadenando la apoptosis en células de glioma. También hemos evaluado los efectos de 1A-116, los cuales mostraron una actividad anti proliferativa y anti invasiva mejorada en este tipo de células64. Actualmente se realizan estudios preclínicos en modelos animales de glioma con resultados prometedores (datos no mostrados). Por otro lado, existe información que vincula a las Rho GTPasas con el incremento del potencial invasivo de células de glioblastoma posterior a la radioterapia. Se ha propuesto la posibilidad de inhibir farmacológicamente las vías de Rac1 con el fin de incrementar la eficacia terapéutica de la radioterapia65-67. Se desarrollaron inhibidores de la interacción de Cdc42 con sus respectivos GEFs. Recientemente se describió a CASIN como un inhibidor de Cdc42 que bloquea la interacción con su GEF intersectina68. Existen otros Inhibidores de Cdc42 más específicos como ML14169 y ZCL27870 que aún se encuentran bajo evaluación.
En otro trabajo, se identificó por screening virtual un inhibidor de RhoA llamado Rhosin, el cual se une al Trp58 de esta proteína y bloquea su actividad afectando el citoesqueleto y la invasión en células de cáncer de mama71.
Inhibición de las interacciones Rho GTPasa-nucleótidos
Otro grupo importante de inhibidores incluye a los compuestos que interfieren de forma específica en la unión de nucleótidos con Rac1. Pocas drogas fueron desarrolladas en esta área, e incluso menos mostraron ser efectivas. Esto se debe a que la afinidad de las GTPasas por el GTP y la concentración intracelular de GTP hace difícil el desarrollo de este tipo de moléculas57.
Uno de los inhibidores más caracterizados de Rac1 es EHT 1864, el cual interviene en la unión de los nucleótidos resultando en la inactivación Rac1. De esta forma se evita el intercambio de nucleótidos mediados por GEF como también la unión de Rac1 con sus efectores, bloqueando efectivamente la transformación mediada por Rac1 constitutivamente activo72.
Recientemente, Arnst y col. presentaron dos inhibidores nuevos denominados #1 y #6, que fueron diseñados para intervenir en el sitio de unión al nucleótido de Rac1. Estos compuestos fueron capaces de bloquear la unión de Rac1 activado con su efector PAK1 de manera concentración-dependiente, sin producir inhibición de Cdc42 o de RhoA73. Estos estudios indican que ambos inhibidores reducen la proliferación celular y la migración en múltiples líneas celulares de cáncer pancreático a concentración micromolares.
Modificación de la regulación espacial de las Rho GTPasas
Las proteínas Rho requieren de una adición post traduccional de residuos lipídicos en sus extremos carboxilos terminales para su translocación a membrana plasmática. En base a esto, se han desarrollado compuestos con la intención de prevenir dicha modificación. En este grupo se encuentran los inhibidores de farnesil-transferasas y de geranil-geranyltransferasas (GGTasa), los cuales demostraron tener un gran efecto antitumoral en la clínica74.
Las estatinas constituyen un grupo de inhibidores que actúan en la vía de HMG CoA reductasa inhibiendo la producción de colesterol y de determinadas proteínas preniladas. De esta forma logran disminuir los niveles de colesterol e inhibir el agregado de los motivos lipídicos necesarios para el correcto funcionamiento de las Rho GTPasas. Esta estrategia cuenta con la desventaja de ser poco selectiva entre las diferentes GTPasas. Sin embargo, existen ensayos clínicos que indican que las estatinas afectan la progresión tumoral75, 76. Datos recientes mostraron un mecanismo de acción independiente de la prenilación, en el cual los efectos de las estatinas son consecuencia de la degradación del pool nuclear de Rac177. Esto afecta la regulación de la progresión del ciclo celular78 y la polimerización de actina en el núcleo79.
De esta manera, promoviendo la degradación de Rac1 nuclear, los efectos de las estatinas pueden ser también una consecuencia directa de la supresión de Rac1 nuclear. Por ejemplo, se puede apreciar un incremento en la expresión de Rac1 nuclear en biopsias de cuello uterino pre malignas, en biopsias con lesiones intraepiteliales escamosas (SIL) de bajo grado y en SIL de alto grado.
También, se observaron altos niveles de Rac1 nuclear en líneas celulares de cáncer de cuello uterino pero no en sus contrapartes normales, sugiriendo un rol por parte de Rac1 nuclear en la progresión de la enfermedad80. También se ha demostrado que la acumulación nuclear de Rac1 regula la invasión tumoral consecuente a un incremento de la señalización de RhoA en el citoplasma.
Otro grupo de compuestos que interfieren con la regulación espacial son los inhibidores de GGtasa I, los cuales inhiben la prenilación proteica. La inhibición de GGtasa I en un modelo murino de cáncer pancreático disminuye el crecimiento tumoral de forma más efectiva que mediante la inhibición de la geranil-geranilación proteica81. Se descubrieron efectos similares en un modelo murino de cáncer pulmonar82.
Inhibición de efectores específicos de las Rho GTPasas
Otra de las estrategias consiste en la inhibición de las proteínas efectoras. Los dos inhibidores de ROCK, Y-27632 y Fasudil se unen al bolsillo de unión al ATP de ROCK1 Y ROCK2 e inhiben la actividad de la serina-treonina-quinasa83,84. Estos compuestos demostraron un efecto interesante en varios tipos tumorales85. Adicionalmente, se probó que el uso de Y-27632 puede disminuir la neurotoxicidad causada por drogas antitumorales como el cisplatino86.
Fasudil fue aprobado en Japón en 1995 para la prevención y tratamiento de vasoespasmo cerebral posterior a cirugía en pacientes con hemorragias subaracnoideas87. Otro inhibidor competitivo de ROCK: Wf-536, demostró inhibir la angiogénesis, el crecimiento tumoral y la metástasis in vivo88.
Debido a que los inhibidores de ROCK no son específicos de isoformas también inhiben otras quinasas de serina/treonina como PKA y PKC a concentraciones altas, por lo cual pueden causar efectos no deseados89. Recientemente se han desarrollado inhibidores selectivos de isoformas de ROCK y se han descripto varias moléculas90. A pesar de que se desarrollaron numerosos inhibidores de ROCK, se reportó un solo inhibidor de ROCK usado en ensayos clínicos para tratamientos en cáncer. Este inhibidor, AT13148, entró a fase I en el 2012 para el tratamiento de tumores sólidos avanzados91.
Inhibición de las Rho GTPasas por otros mecanismos
La azatioprina y su metabolito 6-mercaptopurina son utilizadas como drogas inmunosupresoras. Si bien se los utiliza en la clínica hace tiempo, posteriormente se determinó a Rac1 como un blanco específico de las drogas en los linfocitos T92. Luego se comunicó que ambas drogas eran capaces de disminuir los niveles de Rac1 activo en un modelo murino de carcinoma mamario in vitro93.
Recientemente se descubrió que el agente fitoquímico rocaglamida, que pertenece a la familia de las flavaglinas, inhibe las actividades de Rho, Cdc42 y Rac1 y podría representar una clase de drogas anti tumorales94.
Observaciones finales
La redundancia y plasticidad de las diferentes vías de señalización permiten a las células tumorales adaptarse y superar diferentes dificultades producidas por los tratamientos o el ambiente de origen. Como consecuencia, las terapias dirigidas a un único blanco altamente específico no siempre alcanzan los resultados deseados. Debido a esto surge la necesidad de plantear estrategias con terapias combinadas95. Una opción es la combinación de drogas que modulan múltiples vías de señalización, siendo las más efectivas las que incluyen agentes que inhiben las señales de supervivencia en múltiples vías de traducción de señales.
La inhibición de las Rho GTPasas representa una oportunidad terapéutica interesante. Como se mostró, se han desarrollado varios inhibidores. Mientras que algunos de estos compuestos muestran resultados prometedores, es importante identificar inhibidores más potentes. Sin embargo, los avances en el diseño racional de las drogas junto a un aumento del conocimiento de los mecanismos involucrados en la regulación de la señalización de las Rho GTPasas nos acercan más hacia el desarrollo de inhibidores de Rho GTPasas altamente específicos con potencialidad de aplicación clínica.
Agradecimientos: Julián Maggio es un becario estudiante del Instituto Nacional del Cáncer, Nazareno González es becario doctoral del CONICET, Georgina Cardama es becaria D-TEC de ANPCyT y Daniel Gomez es Investigador Superior del Conicet. Este trabajo ha sido financiado por la Universidad Nacional de Quilmes, ANPCyT y CONICET.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005; 6: 167-80.
2. Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 2007; 29: 356-70.
3. Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992; 70: 401-10.
4. Ellenbroek SI, Collard JG. Rho GTPases: functions and association with cancer. Clin Exp Metastasis 2007; 24: 657-72.
5. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002; 420: 629-35.
6. Cardama GA, Comin MJ, Hornos L, et al. Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anticancer Agents Med Chem 2014; 14: 840-51.
7. Porter AP, Papaioannou A, Malliri A. Deregulation of Rho GTPases in cancer. Small GTPases 2016; 7: 123-38.
8. Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012; 150: 251-63.
9. Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012; 44: 1006-14.
10. Fields AP, Justilien V. The guanine nucleotide exchange factor (GEF) Ect2 is an oncogene in human cancer. Adv Enzyme Regul 2010; 50: 190-200.
11. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 2010; 10: 842-57.
12. Jarzynka MJ, Hu B, Hui K-M, et al. ELMO1 and Dock180, a bipartite Rac1 guanine nucleotide exchange factor, promote human glioma cell invasion. Cancer Res 2007; 67: 7203-11.
13. Wertheimer E, Gutierrez-Uzquiza A, Rosemblit C, Lopez-Haber C, Sosa MS, Kazanietz MG. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell Signal 2012; 24: 353-62.
14. Khosravi-Far R, Solski PA, Clark GJ, Kinch MS, Der CJ. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cel Biol 1995; 15: 6443-53.
15. Qiu RG, Chen J, McCormick F, Symons M. A role for Rho in Ras transformation. Proc Nat Acad Sci USA 1995; 92: 11781-5.
16. Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/Rock signalling and extracellular proteolysis. Nat Cell Biol 2003; 5: 711-9.
17. Wolf K, Mazo I, Leung H, et al. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 2003; 160: 267-77.
18. Nakaya Y, Kuroda S, Katagiri YT, Kaibuchi K, Takahashi Y. Mesenchymal-epithelial transition during somitic segmentation is regulated by differential roles of Cdc42 and Rac1. Dev Cell 2004; 7: 425-38.
19. Lv Z, Hu M, Zhen J, Lin J, Wang Q, Wang R. Rac1/PAK1 signaling promotes epithelial-mesenchymal transition of podocytes in vitro via triggering β-catenin transcriptional activity under high glucose conditions. Int J Biochem Cell Biol 2013; 45: 255-64.
20. Fritz G, Just I, Kaina B. Rho GTPases are over-expressed in human tumors. Int J Cancer 1999; 81: 682-7.
21. Engers R, Ziegler S, Mueller M, Walter A, Willers R, Gabbert HE. Prognostic relevance of increased Rac GTPase expression in prostate carcinomas. Endocr Relat Cancer 2007; 14: 245-56.
22. Lin Y, Zheng Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin Drug Discov 2015; 10: 991-1010.
23. Gomez del Pulgar T, Benitah SA, Valeron PF, Espina C, Lacal JC. Rho GTPase expression in tumourigenesis: evidence for a significant link. Bioessays 2005; 27: 602-13.
24. Liu Y, Song N, Ren K, et al. Expression loss and revivification of RhoB gene in ovary carcinoma carcinogenesis and development. PLoS One 2013; 8: e78417.
25. Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochimi Biophys Acta 2009; 1796: 91-8.
26. Royer C, Lu X. Epithelial cell polarity: A major gatekeeper against cancer? Cell Death Differ 2011; 18: 1470-7.
27. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer 2009; 9: 239-52.
28. Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal 2010; 8: 23.
29. Mackay AR, Gomez DE, Nason AM, Thorgeirsson UP. Studies on the effects of laminin, E-8 fragment of laminin and synthetic laminin peptides PA22-2 and YIGSR on matrix metalloproteinases and tissue inhibitor of metalloproteinase expression. Lab Invest 1994; 70: 800-6.
30. Zavarella S, Nakada M, Belverud S, et al. Role of Rac1-regulated signaling in medulloblastoma invasion. Laboratory investigation. J Neurosurg Pediatr 2009; 4: 97-104.
31. Salhia B, Tran NL, Chan A, et al. The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am J Pathol 2008; 173: 1828-38.
32. Chan AY, Coniglio SJ, Chuang Y-y, et al. Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene 2005; 24: 7821-9.
33. de Lorenzo MS, Ripoll GV, Yoshiji H, et al. Altered tumor angiogenesis and metastasis of B16 melanoma in transgenic mice overexpressing tissue inhibitor of metalloproteinases-1. In Vivo 2003; 17: 45-50.
34. Bryan BA, D’Amore PA. What tangled webs they weave: Rho-GTPase control of angiogenesis. Cell Mol Life Sci 2007; 64: 2053-65.
35. van Leeuwen FN, van der Kammen RA, Habets GG, Collard JG. Oncogenic activity of Tiam1 and Rac1 in NIH3T3 cells. Oncogene 1995; 11: 2215-21.
36. Li Z, Liu Q, Piao J, et al. Clinicopathological implications of Tiam1 overexpression in invasive ductal carcinoma of the breast. BMC Cancer 2016; 16: 681.
37. Razidlo GL, Magnine C, Sletten AC, et al. Targeting pancreatic cancer metastasis by inhibition of Vav1, a driver of tumor cell invasion. Cancer Res 2015; 75: 2907-15.
38. Yang C, Liu Y, Leskow FC, Weaver VM, Kazanietz MG. Rac-GAP-dependent inhibition of breast cancer cell proliferation by {beta}2-chimerin. J Biol Chem 2005; 280: 24363-70.
39. Menna PL, Skilton G, Leskow FC, Alonso DF, Gomez DE, Kazanietz MG. Inhibition of aggressiveness of metastatic mouse mammary carcinoma cells by the beta2-chimaerin GAP domain. Cancer Res 2003; 63: 2284-91.
40. Gomez DE, Armando RG, Alonso DF. AZT as a telomerase inhibitor. Front Oncol 2012; 2: 113.
41. Armando RG, Gomez DM, Gomez DE. Azt exerts its antitumoral effect by telomeric and non-telomeric effects in a mammary adenocarcinoma model. Oncol Rep 2016; 36: 2731-6.
42. Qu Y, Mao M, Li X, et al. Enhanced migration and cxcr4 over-expression in fibroblasts with telomerase reconstitution. Mol Cell Biochem 2008; 313: 45-52.
43. Yeh Y-M, Pan Y-T, Wang T-CV. Cdc42/Rac1 participates in the control of telomerase activity in human nasopharyngeal cancer cells. Cancer Lett 2005; 218: 207-13.
44. Gomez DLM, Armando RG, Cerrudo CS, Ghiringhelli PD, Gomez DE. Telomerase as a cancer target. Development of new molecules. Curr Top Med Chem 2016; 16: 2432-40.
45. Fernández Larrosa PN, Ruíz Grecco M, Mengual Gómez D, et al. Rac3 more than a nuclear receptor coactivator: A key inhibitor of senescence that is downregulated in aging. Cell Death Dis 2015; 6: e1902.
46. Chen PC, Peng JR, Huang L, et al. Overexpression of human telomerase reverse transcriptase promotes the motility and invasiveness of HepG2 cells in vitro. Oncol Rep 2013; 30: 1157-64.
47. Gomez DL, Farina HG, Gomez DE. Telomerase regulation: A key to inhibition? (Review). Int J Oncol 2013; 43: 1351-6.
48. Tyler JJ, Allwood EG, Ayscough KR. WASP family proteins, more than Arp2/3 activators. Biochem Soc Trans 2016; 44: 1339-45.
49. Choi S, Anderson RA. IQGAP1 is a phosphoinositide effector and kinase scaffold. Adv Biol Regul 2016; 60: 29-35.
50. Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev 2009; 28: 51-63.
51. Parsons M, Adams JC. Rac regulates the interaction of fascin with protein kinase c in cell migration. J Cell Sci 2008; 121: 2805-13.
52. Wei L, Surma M, Shi S, Lambert-Cheatham N, Shi J. Novel insights into the roles of Rho kinase in cancer. Arch Immun Ther Exp (Warsz) 2016; 64: 259-78.
53. Rattan S, Singh J. RhoA/ROCK pathway is the major molecular determinant of basal tone in intact human internal anal sphincter. Am J Physiol Gastrointest Liver Physiol 2012; 302: G664-75.
54. Dawson JC, Bruche S, Spence HJ, Braga VMM, Machesky LM. Mtss1 promotes cell-cell junction assembly and stability through the small GTPase Rac1. PLoS One 2012; 7: e31141.
55. Zandvakili I, Lin Y, Morris JC, Zheng Y. Rho GTPases: anti- or pro-neoplastic targets?. Oncogene 2017; 36: 3213-22.
56. Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA 2004; 101: 7618-23.
57. Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov Today 2006; 11: 580-94.
58. Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol 2007; 152: 9-20.
59. Bid HK, Roberts RD, Manchanda PK, Houghton PJ. RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther 2013; 12: 1925-34.
60. Kaneto N, Yokoyama S, Hayakawa Y, Kato S, Sakurai H, Saiki I. Rac1 inhibition as a therapeutic target for gefitinib-resistant non-small-cell lung cancer. Cancer Sci 2014; 105: 788-94.
61. Citterio C, Menacho-Marquez M, Garcia-Escudero R, et al. The Rho exchange factors Vav2 and Vav3 control a lung metastasis-specific transcriptional program in breast cancer cells. Sci Signal 2012; 5: ra71.
62. Montalvo-Ortiz BL, Castillo-Pichardo L, Hernández E, et al. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J Biol Chem 2012; 287: 13228-38.
63. Cardama GA, Comin MJ, Hornos L, et al. Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anticancer Agents Med Chem 2014; 14: 840-51.
64. Gonzalez N, Cardama GA, Comin MJ, et al. Pharmacological inhibition of Rac1-PAK1 axis restores tamoxifen sensitivity in human resistant breast cancer cells. Cell Signal 2017; 30: 154-61.
65. Cardama GA, Gonzalez N, Ciarlantini M, et al. Proapoptotic and antiinvasive activity of Rac1 small molecule inhibitors on malignant glioma cells. Onco Targets Ther 2014; 7: 2021-33.
66. Hwang S-Y, Jung J-W, Jeong J-S, et al. Dominant-negative Rac increases both inherent and ionizing radiation-induced cell migration in C6 rat glioma cells. Int J Cancer 2006; 118: 2056-63.
67. Delmas C, Heliez C, Cohen-Jonathan E, et al. Farnesyltransferase inhibitor, R115777, reverses the resistance of human glioma cell lines to ionizing radiation. Int J Cancer 2002; 100: 43-8.
68. Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem 2007; 282: 35666-78.
69. Florian MC, Dörr K, Niebel A, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 2012; 10: 520-30.
70. Hong L, Kenney SR, Phillips GK, et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J Biol Chem 2013; 288: 8531-43.
71. Friesland A, Zhao Y, Chen Y-H, Wang L, Zhou H, Lu Q. Small molecule targeting Cdc42–intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc Natl Acad Sci USA 2013; 110: 1261-6.
72. Shang X, Marchioni F, Sipes N, et al. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem Biol 2012; 19: 699-710.
73. Arnst JL, Hein AL, Taylor MA, et al. Discovery and characterization of small molecule rac1 inhibitors. Oncotarget 2017; 8: 34586-600.
74. Mazieres J, Pradines A, Favre G. Perspectives on farnesyl transferase inhibitors in cancer therapy. Cancer Lett 2004; 206: 159-67.
75. Farina HG, Bublik DR, Alonso DF, Gomez DE. Lovastatin alters cytoskeleton organization and inhibits experimental metastasis of mammary carcinoma cells. Clin Exp Metastasis 2002; 19: 551-9.
76. Chan KKW, Oza AM, Siu LL. The statins as anticancer agents. Clin Cancer Res 2003; 9: 10-9.
77. Tanaka S, Fukumoto Y, Nochioka K, et al. Statins exert the pleiotropic effects through small GTP-binding protein dissociation stimulator upregulation with a resultant Rac1 degradation. Arterioscler Thromb Vasc Biol 2013; 33: 1591-600.
78. Michaelson D, Abidi W, Guardavaccaro D, et al. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J Cell Biol 2008; 181: 485-96.
79. Navarro-Lérida I, Pellinen T, Sanchez Susana A, et al. Rac1 nucleocytoplasmic shuttling drives nuclear shape changes and tumor invasion. Developl Cell 2015; 32: 318-34.
80. Mendoza-Catalán MA, Cristóbal-Mondragón GR, Adame-Gómez J, et al. Nuclear expression of Rac1 in cervical premalignant lesions and cervical cancer cells. BMC Cancer 2012; 12: 116.
81. Lu J, Chan L, Fiji HDG, Dahl R, Kwon O, Tamanoi F. In vivo antitumor effect of a novel inhibitor of protein geranylgeranyltransferase-I. Mol Cancer Ther 2009; 8: 1218-26.
82. Zimonjic DB, Chan LN, Tripathi V, et al. In vitro and in vivo effects of geranylgeranyltransferase I inhibitor P61A6 on non-small cell lung cancer cells. BMC Cancer 2013; 13: 198.
83. Nagumo H, Sasaki Y, Ono Y, Okamoto H, Seto M, Takuwa Y. Rho kinase inhibitor HA-1077 prevents Rho-mediated myosin phosphatase inhibition in smooth muscle cells. Am J Physiol Cell Physiol 2000; 278: C57-65.
84. Uehata M, Ishizaki T, Satoh H, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997; 389: 990-4.
85. Kale VP, Hengst JA, Desai DH, Amin SG, Yun JK. The regulatory roles of ROCK and MRCK kinases in the plasticity of cancer cell migration. Cancer Lett 2015; 361: 185-96.
86. James SE, Burden H, Burgess R, et al. Anti-cancer drug induced neurotoxicity and identification of Rho pathway signaling modulators as potential neuroprotectants. Neurotoxicol 2008; 29: 605-12.
87. Dong M, Yan BP, Liao JK, Lam Y-Y, Yip GWK, Yu C-M. Rho-kinase inhibition: a novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov Today 2010; 15: 622-9.
88. Fritz G, Kaina B. Rho GTPases: promising cellular targets for novel anticancer drugs. Curr Cancer Drug Targets 2006; 6: 1-14.
89. Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J 2007; 408: 297-315.
90. Boerma M, Fu Q, Wang J, et al. Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of Rho kinase or atorvastatin. Blood Coagul Fibrinolysis 2008; 19: 709-18.
91. Sadok A, McCarthy A, Caldwell J, et al. Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res 2015; 75: 2272-84.
92. Tiede I, Fritz G, Strand S, et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4(+) T lymphocytes. J Clin Invest 2003; 111: 1133-45.
93. Menna PL, Parera RL, Cardama GA, Alonso DF, Gomez DE, Farina HG. Enhanced cytostatic activity of statins in mouse mammary carcinoma cells overexpressing Beta2-chimaerin. Mol Med Rep 2009; 2: 97-102.
94. Becker MS, Müller PM, Bajorat J, et al. The anticancer phytochemical rocaglamide inhibits Rho GTPase activity and cancer cell migration. Oncotarget 2016; 7: 51908-21.
95. Dent P, Curiel DT, Fisher PB, Grant S. Synergistic combinations of signaling pathway inhibitors: mechanisms for improved cancer therapy. Drug Resist Updat 2009; 12: 65-73.