
SERGIO LAURITO, MARÍA ROQUÉ
Instituto de Histología y Embriología (IHEM), Facultad de Ciencias Exactas y Naturales, Universidad Nacional de Cuyo – CONICET, Mendoza, Argentina
Resumen La región q11-q13 del cromosoma 15 humano es proclive a sufrir alteraciones genéticas. Algunos
genes de la región presentan expresión parental diferencial monoalélica, regulada por imprinting (EI). Errores en la regulación del EI, disomías uniparentales (DSU), así como también el cambio en el número de copias genómicas (CNV) producidos por sitios susceptibles de quiebre cromosómico (BP), producen alteraciones en esta región. Las enfermedades más frecuentes asociadas son el síndrome de Prader-Willi, el síndrome de Angelman y el síndrome de microduplicación 15q11-q13. En el presente trabajo analizamos la región 15q11-q13 por Methyl specific-multiplex ligation-dependent probe amplification (MS-MLPA) en 181 muestras de ADN derivadas a nuestro servicio de análisis genético molecular. En este trabajo mostramos que, de las 181 muestras, 39 presentaron alteraciones detectables por MS-MLPA. El 61.5% (24/39) de esas alteraciones detectadas fueron deleciones, el 5.1% (2/39) duplicaciones y el 33.3%(13/39) DSU/EI. Los CNV fueron 4 veces más frecuentes que las DSU/EI (OR = 4; IC 95%: 1.56-10.25) consistente con la literatura. Entre los CNV, dos casos atípicos permiten postular posibles sitios BP que no han sido informados en la literatura previamente.
Palabras clave: Síndrome de Prader-Willi, síndrome de Angelman, MS-MLPA, sitios de quiebre cromosómico
Abstract Variation analysis of the number of copies and methylene patterns in region 15q11-q13. Hu-
man chromosome 15q11-q13 region is prone to suffer genetic alterations. Some genes of this region have a differential monoallelic imprinting-regulated expression pattern. Defects in imprinting regulation (IE), uniparental disomy (UPD) or copy number variation (CNV) due to chromosomal breakpoints (BP) in 15q11-q13 region, are associated with several diseases. The most frequent are Prader-Willi syndrome, Angelman syndrome and 15q11-q13 microduplication syndrome. In this work, we analyzed DNA samples from 181 patients with phenotypes which were compatible with the above-mentioned diseases, using Methyl specific-multiplex ligation-dependent probe amplification (MS-MLPA). We show that, of the 181 samples, 39 presented alterations detectable by MS-MLPA. Of those alterations, 61.5% (24/39) were deletions, 5.1% (2/39) duplications and 33.3% (13/39) UPD/IE. The CNV cases were 4 times more frequent than UPD/IE (OR= 4; IC 95%: 1.56-10.25), consistent with the literature. Among the CNVs, two atypical cases allow to postulate new possible BP sites that have not been reported previously in the literature.
Key words: Prader-Willi syndrome, Angelman syndrome, MS-MLPA, chromosome breakpoint
Recibido: 2-VI-2017 Aceptado: 21-XI-2017
Dirección postal: Dra. María Roqué, Instituto de Histología y Embriología, (IHEM), Universidad Nacional de Cuyo – CONICET, Av. Ruiz Leal s/n, Parque General San Martín, 5500 Mendoza, Argentina
e-mail: mroque@mendoza-conicet.gob.ar
Las alteraciones genéticas y epigenéticas producidas en la región cromosómica 15q11.2-q13 son la causa de tres afecciones bien caracterizadas: síndrome de Prader-Willi (PWS, OMIM #176270), síndrome de Angelman (AS, OMIM #105830) y síndrome de microduplicación 15q11-q13 (MDS15q11-q13, OMIM #608636). Las principales características clínicas de PWS son hipotonía grave, hipogonadismo, trastornos alimentarios y obesidad mórbida en la infancia temprana, y se presenta con una frecuencia de 1:25 000 nacidos1, 2. Los individuos con fenotipo AS poseen generalmente retraso en el desarrollo, trastornos motores, trastornos del habla y de comunicación en general, episodios de risa y ataxia. Se estima su incidencia en 1:30 000 nacidos2-4”. Por otro lado, MDS 15q11-13 produce hipotonía, retraso en el desarrollo, retraso motor y trastornos del espectro autista, y se presenta en 1:30 000 nacimientos2, 5.
La expresión de los genes de la región cromosómica 15q11-q13 sometidos a imprinting es monoalélica, es decir que solo se expresa una de las copias ubicada cada una en uno de los dos cromosomas homólogos. Dependiendo del gen, la expresión es materna o paterna6, 7 (Fig. 1). En el caso de la región 15q11-q13, en células normales se expresan los genes paternos SNRPN y NDN y quedan silenciados los maternos8. En el caso del gen UBE3A,
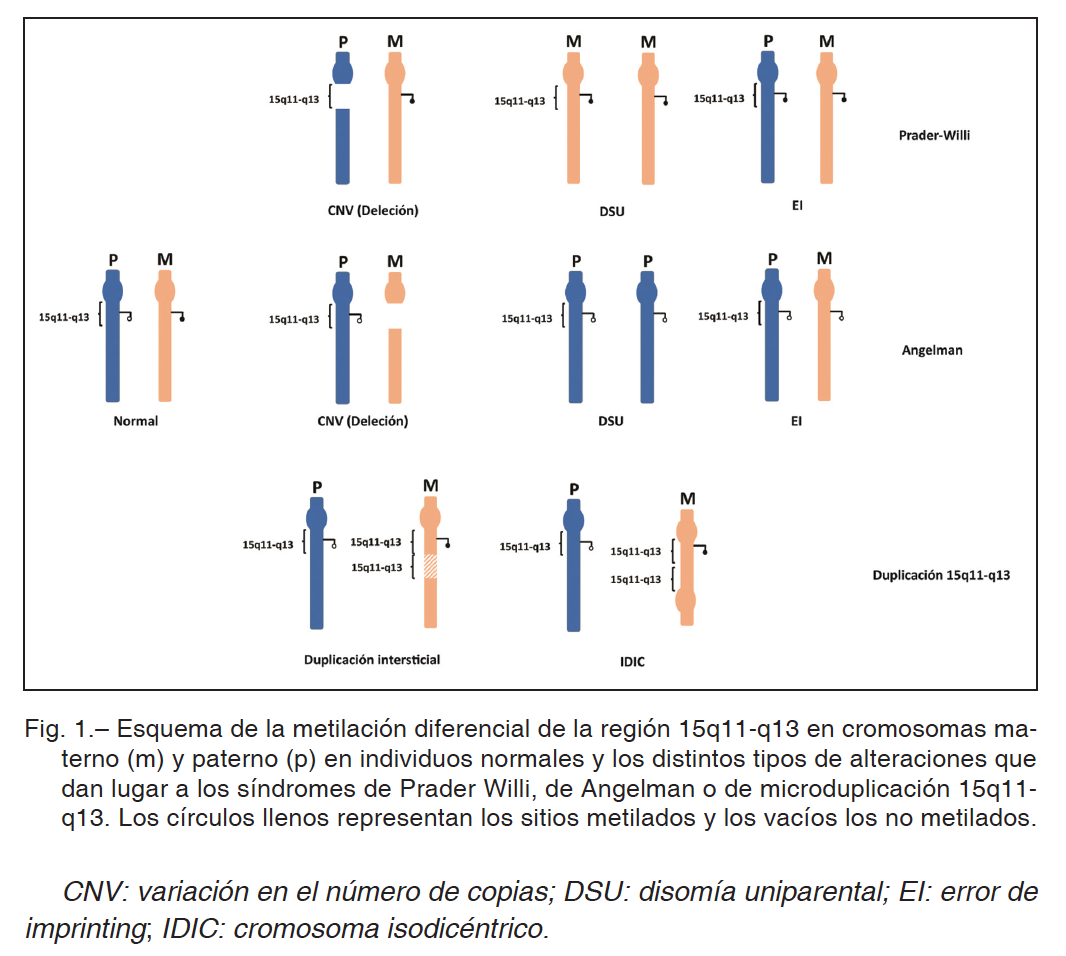
situado en la misma región, la situación es inversa: se expresa la copia cromosómica materna y permanece silenciada la copia paterna. Esta regulación de expresión se produce por metilación de ADN en una secuencia específica de la región cromosómica denominada centro de imprinting (IC, del inglés imprinting center) que afecta a la expresión de los genes SNRPN, NDN y UBE3A6, 9 . Cabe mencionar que la expresión de UBE3A regulada por imprinting sólo ocurre en células del cerebro10.
Las deleciones que ocurren en esta región, (denominada “región crítica de PWS/AS” o PWACR), producen fenotipos PWS o AS4, 11 (Fig. 1), mientras que duplicaciones producen fenotipos MD15q11-q135. La alteración en los patrones normales de metilación también se asocia con PWS y AS, aunque en este caso las características clínicas son menos graves12. La metilación aberrante de las copias paternas de los genes SNRPN y NDN da como resultado final que el 100% de los alelos se encuentren metilados (hipermetilación), asociándose con PWS. En cambio, una des-metilación aberrante de las copias maternas de los mismos genes genera un 0% de alelos metilados (hipometilación), asociándose con AS.
Los mecanismos por los cuales se produce un desbalance en la cantidad de copias metiladas son varios, pudiéndose dividir en dos grupos: 1. los que se producen por una variación en el número de copias (CNV, del inglés copy number variation), que incluyen deleciones, duplicaciones intersticiales, translocaciones cromosómicas y formación de cromosomas isodicéntricos; 2. los que se producen por una disomía uniparental (DSU) o por errores en el centro de imprinting (EI)13.
La región 15q11-q13 es proclive a sufrir CNV debido a que presenta 5 puntos de quiebre canónicos (del inglés breaking points o BP). Estos sitios BP canónicos están integrados por el gen HERC2 (correspondiente al BP3), los pseudogenes de HERC2 (correspondientes a los BP1, BP2, BP4 y BP5), y a repeticiones de bajo número de copias (LCR)14. Debido a la homología entre estas secuencias, un mal alineamiento entre los LCR de cromosomas homólogos produce una recombinación no alélica entre ellos durante la meiosis, lo que resulta en una pérdida o ganancia de material genético. En las deleciones, en el caso en que se pierda la copia materna (SNRPN y NDN metilados, UBE3A desmetilado) y solo quede la copia paterna (SNRPN y NDN desmetilados y UBE3A metilado), se produce AS. Por el contrario, si se pierde el alelo paterno, ocurre la situación inversa y se produce PWS. Los métodos de detección de CNV incluyen hibridación fluorescente in situ, análisis de microarreglos cromosómicos y secuenciación automática10, 15.
En otros casos, el patrón de metilación puede ser alterado al heredarse dos cromosomas del mismo origen parental (DSU), error que suele producirse durante la segregación de cromosomas durante la anafase de la meiosis I. Dos cromosomas maternos (metilados) se asociarán con un fenotipo PWS mientras que dos cromosomas paternos (des-metilados) con AS. Y finalmente, un EI puede producir una metilación aberrante en el cromosoma paterno (compatible con fenotipo PWS), o una des-metilación aberrante del cromosoma materno (compatible con fenotipo AS). Para detectar DSU/EI se suele utilizar PCR metil-específica o secuenciación metil-específica. Para detectar ambas en forma combinada (tanto CNV como DSU/EI) se requiere utilizar diferentes metodologías cualitativas y cuantitativas.
La metodología Methyl specific multiplex ligation-dependent probe amplification (MS-MLPA) presenta una gran ventaja para estas determinaciones, al permitir detectar CNV y DSU/EI de forma simultánea en un mismo análisis16, 17.
Las muestras incluidas en este trabajo fueron derivadas desde diferentes centros diagnósticos del país al servicio de análisis genético molecular de nuestra institución (STAN, IHEM Mendoza), acompañadas de un pedido médico para el análisis de la región 15q11-q13 en 181 pacientes con sospecha clínica de PWS/AS/MD. En este estudio presentamos los resultados que permitieron obtener los análisis por MS-MLPA en muestras de ADN de leucocitos, y mostramos un análisis de frecuencias y diferencias estadísticas entre los distintos tipos de alteraciones encontradas y las diferencias con la literatura publicada.
Materiales y métodos
Entre los años 2011 y 2016 se incluyeron 181 muestras de sangre derivadas a nuestro Instituto para determinar alteraciones de metilación y de número de copias en la región cromosómica 15q11-q13. Los pacientes fueron derivados por médicos genetistas de distintos centros diagnósticos de Argentina, con fenotipos compatibles con los síndromes de PWS, AS y MD 15q11-q13. El estudio no incluye datos de la historia clínica de los pacientes, debido a que se recibieron únicamente muestras de sangre periférica para la obtención de ADN.
Se realizó la extracción de ADN por método convencional basado en CTAB (bromuro de hexadeciltimetilamonio). Brevemente, se extrajeron los linfocitos totales de 3 ml de sangre de cada paciente utilizando buffer T10E10 (Tris base 10 mM, EDTA 10 mM) y centrifugaciones sucesivas hasta obtener un pellet de linfocitos limpio. A continuación el pellet fue tratado con buffer CTAB (2% CTAB, 0.2 % β-mercaptoetanol, NaCl 1.4 M, EDTA 20 mM y Tris 10 mM) y temperatura (60 °C) durante 60 minutos. Luego, el ADN fue purificado utilizando cloroformo: alcohol isoamílico (24:1) y precipitado con etanol 100%.
La metodología aplicada para el análisis de metilación y de número de copias fue MS-MLPA utilizando el kit ME028 (MRC-Holland). Esta técnica se basa en la hibridación de pares de sondas en sitios de ADN adyacentes, y su posterior ligación e incubación con una enzima metil-sensible. Estas enzimas digieren solo los fragmentos no metilados. Luego, una reacción por PCR con primers fluorescentes permite amplificar las sondas no digeridas, o sea las que hibridaron a sitios de ADN metilado. Finalmente, los productos de PCR obtenidos fueron separados por electroforesis capilar. Los electroferogramas resultantes fueron analizados utilizando el programa informático GeneMarker v1.75 (Softgenetics). Los ensayos se llevaron a cabo según recomendaciones del fabricante, incluyendo algunas modificaciones puestas a punto en nuestro laboratorio como previamente publicamos18.
Resultados
Entre las 181 muestras analizadas, 39 (21.5%) presentaron alteraciones detectables por MS-MLPA. En 24/39 (61.5%) se hallaron deleciones del alelo materno o paterno, 2/39 (5.1%) presentaron duplicaciones y 13/39 (33.3%) DSU/EI. De esto surge que la alteración más frecuente detectada es la CNV (26/39, 66.6%), que difiere significativamente (p < 0.05) de la frecuencia de los otros tipos de alteraciones detectadas (DSU/EI) (33.3%). Análisis de odds ratio (OR) revelan que las CNV ocurren 4 veces más frecuentemente que las DSU/EI (OR = 4; IC 95%: 1.56-10.25).
Las 26 CNV abarcan distintos genes de la región 15q11-q13 que son representados en la Fig. 2. La extensión de las 24 deleciones se muestra en color rojo y la de las 2 duplicaciones en color verde (negro y gris respectivamente, en la versión impresa). Entre las CNV, las deleciones son las más frecuentes (24/26, 92%) e involucran diferentes sitios BP (Fig. 2). Según el International Standards for Cytogenomic Arrays consortium (ISCA) existen dos tipos de deleciones típicas de la región 15q11-q13: la deleción clase I, comprendida entre BP1 y BP3 y la deleción clase II, entre BP2 a BP3. Esta última deleción clase II es considerada la región crítica en los síndromes de PWS y AS y, consistente con esto, la mayoría de las deleciones detectadas en este trabajo (17/24, 70.8%) se extienden entre estos BP2 y BP3. Sin embargo, es interesante resaltar que 5/24 (20.8%) abarcaron una región alternativa de mayor extensión, desde BP1 a BP3 (deleción clase I).
Cabe destacar dos casos de deleciones en los cuales las regiones involucradas fueron atípicas. En un caso se encontró una deleción entre BP2-BP3 sin abarcar la región de forma completa, ya que el gen MKRN3 situado próximo a BP2 no estaba delecionado. Esto puede sugerir la presencia de un sitio de quiebre alternativo entre BP2 y BP3. En el segundo caso atípico, la deleción ocurrió en sitios de quiebre no canónicos ubicados entre BP2-BP3 y BP3-BP4 afectando a genes de ambos sectores, i.e.: SNRPN, UBE3A, ATP10A y GABRB3 del sector BP2-BP3 y APBA2 del sector BP3-BP4. Esta observación, nuevamente, sugiere la presencia de posibles sitios de quiebre cromosómicos alternativos entre los BPs conocidos (Fig. 2).

De las 26 CNV identificadas, 2 correspondieron a duplicaciones. Ambas duplicaciones intersticiales, si bien según la literatura son mucho menos frecuentes que las deleciones2, abarcan la misma región cromosómica que éstas, desde el BP1 al BP4. Los genes involucrados son NIPA1, TUBGCP5 (entre BP1-BP2), MKRN3, MAGEL2, NDN, SNRPN, UBE3A, ATP10A, GABRB3 (entre BP2-BP3) y APBA2 (entre BP3-BP4).
Finalmente, en 13 de 39 muestras (33.3%) se detectó una alteración del perfil de metilación por MS-MLPA sin incluir CNV, infiriéndose como causa DSU o EI.
Discusión
En nuestros resultados observamos una frecuencia significativamente mayor de CNV que de DSU/EI en la población de pacientes con alteraciones en 15q11-q13. Esto se encuentra en línea con los hallazgos previamente publicados en otros trabajos, en los cuales se observa que la frecuencia de CNV es 65%-75%, mientras que para DSU/EI es 10%-30%12. Según la literatura, la DSU es más frecuente que EI en el caso de PWS (25%-30% vs. 1%), mientras que para AS la diferencia entre ambos tipos de alteraciones es menor (7.0% vs. 2.5%)22, 23. De todas maneras, en nuestro estudio no se pudo diferenciar entre estas dos causas, debido a que MS-MLPA no está diseñada para identificar el origen parental de los cromosomas, por lo que no posibilita diferenciar ente DSU y EI.
De las muestras analizadas surge el dato llamativo de que, entre las CNV detectadas, las deleciones son mucho más frecuentes que las duplicaciones (92% vs. 8%). Nuestros datos difieren de lo registrado en estudios de otras poblaciones, donde la frecuencia de deleciones y duplicaciones son similares2. No tenemos explicación biológica para esta diferencia, y estimamos que podría deberse a un sesgo de las muestras recibidas en nuestro servicio. Es posible que se deriven más frecuentemente muestras para análisis de PWS/AS (asociados en su mayoría –> 60%– a deleciones) y no para trastornos del espectro autista u otros síntomas diferentes de los observados en PWS o AS2, 5 (asociados mayormente a duplicaciones).
Con respecto a los sitios de quiebre involucrados, en algunos casos pudimos encontrar que las deleciones habían ocurrido entre BPs que no han sido mencionados previamente por otros13, 19. Es posible proponer para estos casos atípicos la existencia de otros BPs20 que hayan provocado deleciones con características clínico-patológicas similares a PWS/AS.
De las muestras analizadas en nuestro servicio, el porcentaje de detección de alteraciones fue de un 21.5%, equivalente a una tasa aproximada de 1:4. Una búsqueda bibliográfica revela que más del 99% de los casos de PWS y el 80% de los de AS son detectables por MS-MLPA1. En los casos de AS no detectables por MS-MLPA, un 11% está asociado a variantes patológicas del gen UBE3A y un 9% a microdeleciones en el centro de imprinting de la región 15q1115. Esto indica que MS-MLPA tiene una alta sensibilidad y especificidad para detectar alteraciones de PWS/AS. Por esto, suponemos que la tasa de detección más baja en la población analizada en este trabajo se puede deber a que las 181 muestras correspondan a pacientes sin certeza diagnóstica. A su vez, cabe resaltar que los rasgos fenotípicos de PWS/AS son solapables con otros síndromes, ej. craneofaringioma1,19 distrofia miotónica tipo I, diferentes neuropatías incluyendo atrofia medular espinal21. Por lo tanto, consideramos que nuestros datos revelan la importancia de corroborar el diagnóstico clínico de PWS/AS por MS-MLPA, dada la complejidad de un diagnóstico certero basado sólo en el criterio clínico-patológico.
En conclusión, la metodología MS-MLPA ha permitido identificar casos con PWS/AS de forma rápida y con costos razonables. Pudo establecerse el diagnóstico, diferenciar entre las causas de CNV o DSU/EI e identificar el tamaño de los CNV. Un análisis global ha permitido determinar que tipo de alteraciones ocurren más frecuentemente en nuestra población. A su vez, ha permitido postular posibles sitios de quiebre cromosómicos que no han sido reportados en la literatura previamente.
Conflicto de intereses: Ninguno para declarar
Bibliografía
1. Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med 2012; 14: 10-26.
2. Kalsner L, Chamberlain SJ. Prader-Willi, Angelman, and 15q11-q13 duplication syndromes. Pediatr Clin North Am 2015; 62: 587-606.
3. Dagli AI, Williams CA. Angelman Syndrome. En: https://www.ncbi.nlm.nih.gov/books/NBK1144; consultado mayo 2017
4. Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics 2015; 12: 641-50.
5. Finucane BM, Lusk L, Arkilo D, et al. 15q Duplication syndrome and related disorders. En: https://www.ncbi.nlm.nih.gov/books/NBK367946; consultado mayo 2017
6. Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet Part A 2008; 146: 2041-52.
7. Dawson AJ, Cox J, Hovanes K, Spriggs E. PWS/AS MS-MLPA confirms maternal origin of 15q11.2 microduplication. Case Rep Genet 2015; 2015: 1-3.
8. Bittel DC, Kibiryeva N, Talebizadeh Z, Butler MG. Microarray analysis of gene/transcript expression in Prader-Willi syndrome: deletion versus UPD. J Med Genet 2003; 40: 568-74.
9. Perk J, Makedonski K, Lande L, Cedar H, Razin A, Shemer R. The imprinting mechanism of the Prader-Willi/Angelman regional control center. EMBO J 2002; 21: 5807-14.
10. Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med Genet 2010; 70: 1-11.
11. Libov A, Maino M. D. Prader-Willi Syndrome. J Am Optom Assoc 1994; 65: 355-9.
12. Tan WH, Bacino CA, Skinner SA, et al. Angelman syndrome: Mutations influence features in early childhood. Am J Med Genet 2011; 155: 81-90.
13. Depienne C, Moreno-De-Luca D, Heron D, et al. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry 2009; 66: 349-59.
14. Chai JH, Locke DP, Greally JM, et al. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet 2003; 73: 898-925.
15. Buiting K, Cassidy SB, Driscoll DJ, et al. Clinical utility gene card for: Prader-Willi Syndrome. Eur J Hum Genet 2014; 22.
16. Nygren AOH, Ameziane N, Duarte HMB, et al. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res 2005; 33: 1-9.
17. Procter M, Chou LS, Tang W, Jama M, Mao R. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin Chem 2006; 52: 1276-83.
18. Branham MT, Marzese DM, Laurito SR, et al. Methylation profile of triple-negative breast carcinomas. Oncogenesis 2012; 1: 1-7.
19. Goldstone AP, Patterson M, Kalingag N, et al. Fasting and postprandial hyperghrelinemia in Prader-Willi syndrome Is partially explained by hypoinsulinemia, and is not due to peptide YY 3-36 deficiency or seen in hypothalamic obesity due to craniopharyngioma. J Clin Endocrinol Metab 2005; 90 : 2681-90.
20. Takano K, Lyons M, Moyes C, Jones J, Schwartz C. Two percent of patients suspected of having Angelman syndrome have TCF4 mutations. Clin Genet 2010; 78: 282-8.
21. Cassidy SB, Dykens E, Williams CA. Prader-Willi and Angelman syndromes: sister imprinted disorders. Am J Med Genet 2000; 97: 136-46.
22. Robinson WP, Christian SL, Kuchinka BD, et al. Somatic segregation errors predominantly contribute to the gain or loss of a paternal chromosome leading to uniparental disomy for chromosome 15. Clin Genet 2000; 57: 349-58.
23. Varela MC, Kok F, Otto PA, Koiffmann CP. Phenotypic variability in Angelman syndrome: comparison among different deletion classes and between deletion and UPD subjects. Eur J Hum Genet 2004;12 : 987-92.
24. Wandstrat AE, Leana-Cox J, Jenkins L, Schwartz S. Molecular cytogenetic evidence for a common breakpoint in the largest inverted duplications of chromosome 15. Am J Hum Genet 1998; 62: 925-36.
25. Henkhaus RS, Kim S-J, Kimonis VE, et al. Methylation-specific multiplex ligation-dependent probe amplification and identification of deletion genetic subtypes in Prader-Willi syndrome. Genet Test Mol Biomarkers 2012; 16: 178-86.